Bioinformatik: Eine Anleitung zum Lesen der Gene
|
|
|
- Heike Fleischer
- vor 8 Jahren
- Abrufe
Transkript
1 Bioinformatik: Eine Anleitung zum Lesen der Gene Gerhard Thallinger Institut für Genomik und Bioinformatik, TU Graz 1 1

2 Themen für Projekte, Bachelor- und Masterarbeiten Themen sind zu finden unter Für weitere Themen und eigene Ideen kontaktieren Sie 2 3

3 Bioinformatik Gene Funktion 3 5

4 Vorlesungsüberblick Biologische Datenbanken: Überblick Suche Definition und Aufbau Prediktive Methoden basierend auf DNA und Protein Sequenzen Multiples Sequenzalignment Phylogenentische Analyse Next Generation Sequencing Technologien Anwendungen 4 6

5 Biologische Datenbanken 3 große Zentren: National Center for Biotechnology Information (NCBI, NIH, Bethesda, USA) European Bioinformatics Institute (EBI, EMBL, Hinxton, UK) DNA Databank of Japan (DDBJ, NIG, Mishima, Japan) Sehr große Zahl von spezialisierten Datenbanken (SwissProt, KEGG, PDB, PFAM, RFAM, mirbase, ) Jährliche Publikation in der Datenbankausgabe von Nucleic Acids Research 5 7
 Jährliche Publikation in der Datenbankausgabe von Nucleic Acids Research 5")
6 Biologische Datenbanken: Suche basierend auf David Landsman Computational Biology Branch NCBI 6 8

7 Überblick NCBI Entrez System 7 9

8 NCBI: Wieviel Information ist gespeichert? Nucleotide records 167,295,840 Nucleotides 154,192,921,011 Protein sequences 97,127,736 3D structures (PDB) 94,715 Expression data samples (GEO) 1,010,406 Human Unigene clusters 130,029 Complete genomes (GOLD) 17,481 Different taxonomy nodes dbsnp (human only) Human Refgene records PubMed records OMIM records 1,075, ,952,851 40,453 23,175,261 23,163 (Stand August 2013) 8 10
 17,481 Different taxonomy nodes dbsnp (human only) Human Refgene records PubMed records OMIM")
9 Entwicklung des Datenaufkommens 9 11

10
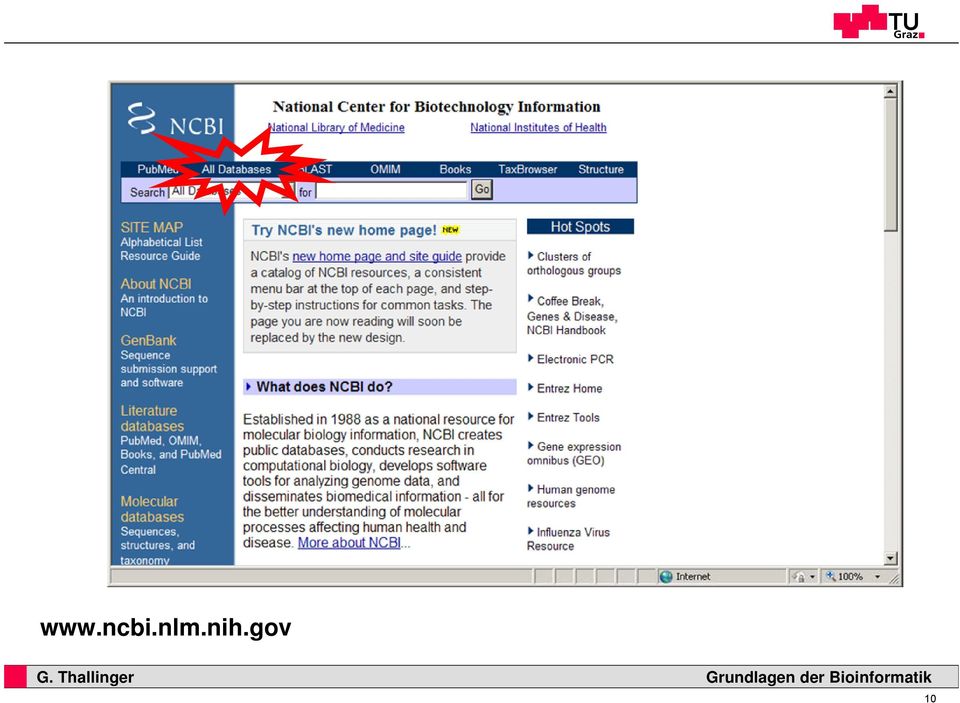
11

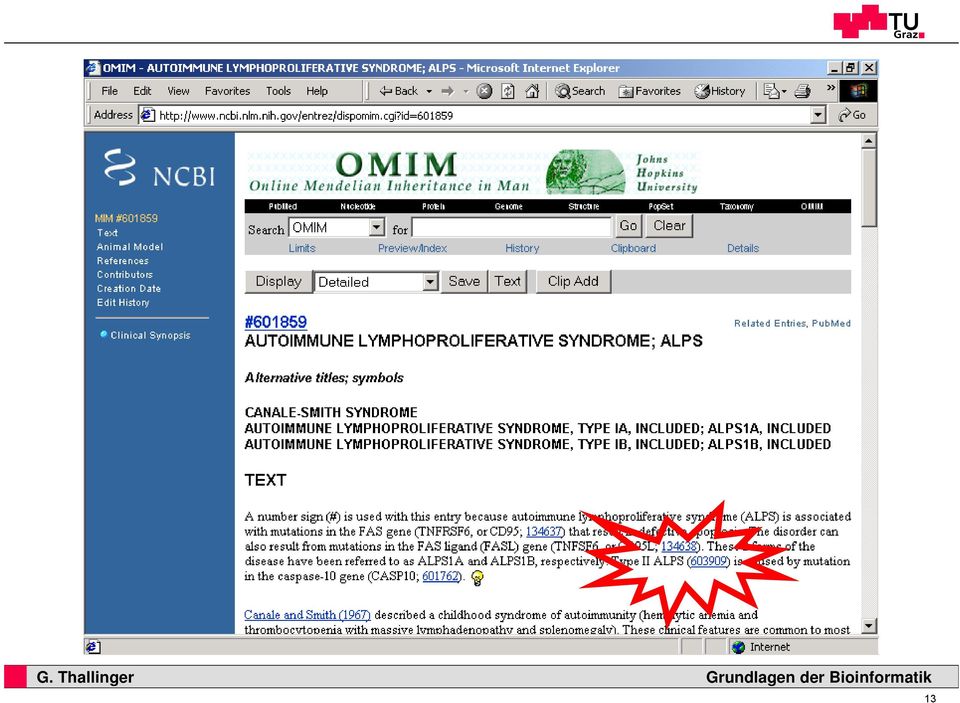
12 Autoimmune lymphoproliferative syndrome 12 14

13 13 15
14 14 16

15 15 17
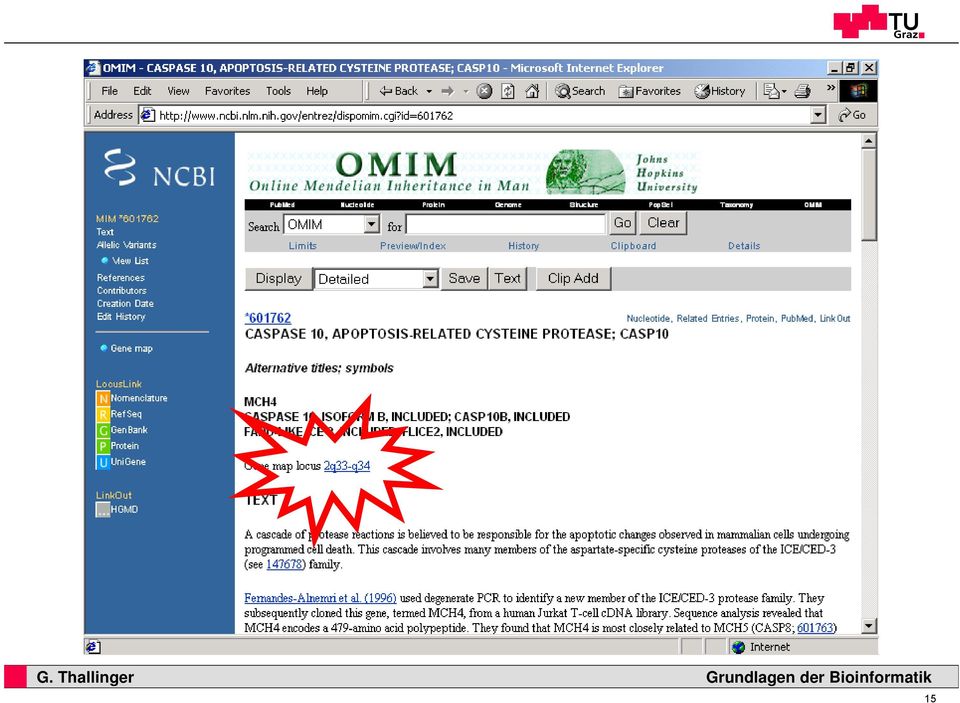
16 16 18

17 17 19

18 18 20

19 19 21

20 20 22
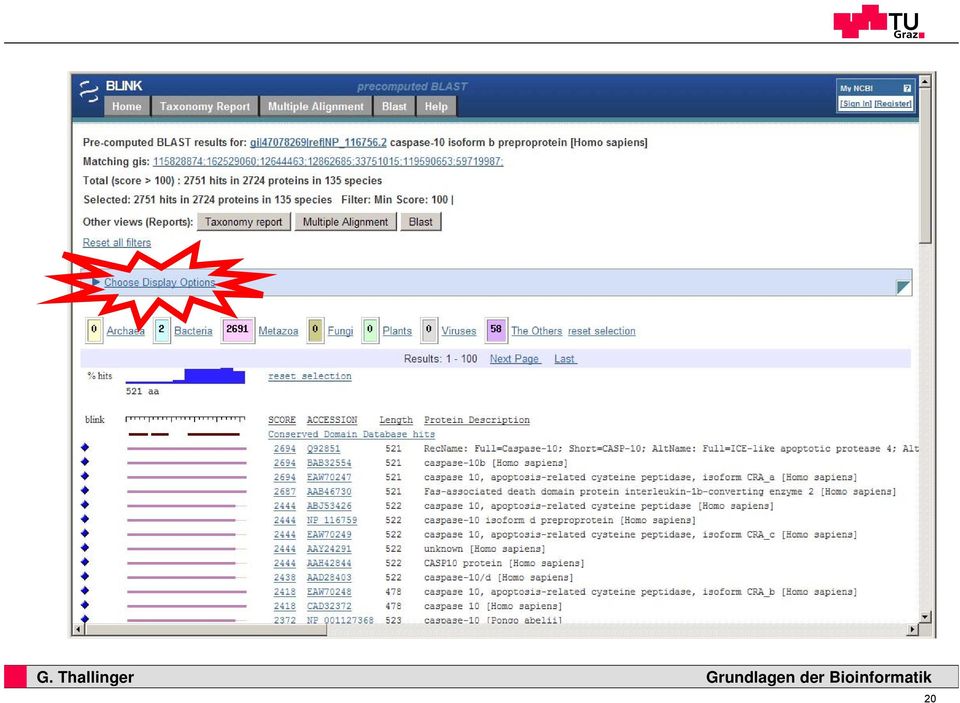
21 21 23
22 22 24
23 23 25
24 24 26
25 ALPS Fallstudie (online) 25 27
26 ALPS Fallstudie (online) 26 31
27 MyNCBI 27 32
28 28 33
29 29 34
30 30 35
31 31 36
32 32 37
33 Biologische Datenbanken: Mehr zur die Suche 33 38
34 34 39
35 35 40
36 36 41
37 37 42
38 38 43
39 39 44
40 Struktursuche
41 41 46
42 42 47
43 43 48
44 44 49
45 45 50
46 Das Ensembl Projekt Wurde initiiert, um Genome von Metazoen zur Verfügung zu stellen Ziel: Die beste automatische Genom-Annotation zu entwickeln Die Kerngruppe analysiert das humane und das Mausgenom Software, Daten und Resultate sind frei verfügbar Aufteilung zwischen EBI und Sanger (vom Wellcome Trust finanziert) Das größte Computerzentrum für die Biologie in Europa Entwicklergemeinschaft > 300 Personen (inkl. beteiligte Firmen) Sanger Center, Hinxton, UK 46 51
47 Das Ensembl Projekt Core database Normalisiert Jeder Datenpunkt genau einmal gespeichert (keine Redundanz) Rasche Aktualisierung Geringster Speicherbedarf ABER: Viele Tabellen Viele JOINS in komplexen Abfragen Sehr langsam für das Data Mining Mart database (EnsMart) Nicht normalisiert Tabellen mit redundant Informationen Optimiert für Abfragen Schnell und flexibel 47 52
48 Vergleichende Genomik in Ensembl Die Compara Datenbank ist eine große Multispecies Datenbank Gen Ortholog / Paralog Vorhersage Protein Clustering Alignments über ganze Genome Berechnung von Synteny Regions 48 53
49 EB-eye
50 Das Ensembl Projekt 50 55
51 Ensembl Core vs. EnsMart Species Data size (Gb) Homo sapiens Mus musculus 92.7 Sus scrofa 25.9 Bos taurus 20.8 Rattus norvegicus 17.9 Pongo abelii 15.7 Gallus gallus 15.4 Canis familiaris 13.7 Mustela putorius 13.7 Pan troglodytes 12.6 Other (53) Total Multi-species Data size (Gb) Ancestral 35.9 Archive 0 Compara Mart Ontology 0.2 Production 0 Stable 2.2 Website 0.1 Total
52 Literatursuche
53 PubMed PubMed ist die Literaturdatenbank der National Library of Medicine PubMed ist aus MEDLINE (CD-ROM Datenbank) hervorgegangen Enthält Publikationen aus dem Gebiet der Medizin und angrenzenden Wissenschaften mit Autoren Journal Zusammenfassung Schlüsselbegriffe Links zu Volltext (beim Verlag) und anderen (NCBI) Datenbanken
54 PubMed: Was ist NICHT enthalten Alle Journale Meeting Abstracts Bücher und Buchkapitel sind nicht indiziert Volltext zu den Publikationen (im Allgemeinen) Teilweise (Open Access Journale, Publikationen aus NIH geförderten Projekten) 54 60
55 55 61
56 PubMed: Beispiele zur Suche Autoren: Autor und Begriff ubject.html Einfache Begriffssuche Suche nach einem Journal Benachrichtigungen
57 PubMed: Die wichtigsten Feldschlüssel [au] Autor [ta] Abkürzung des Journalnamens (journal title abbreviation) [ti] Titel der Publikation [1au] Erstautor (1st author) [dp] Datum der Veröffentlichung (date of publication) [ab] Kurzfassung (abstract) [mh] MeSH Begriff (Medical subject headings term) Überblick: part=pubmedhelp#pubmedhelp.search_field_descrip 57 63
58 Weitere Datenbanken
59 ExPASy 59 65
60 UniprotKB 60 66
61 Prosite - Motivdatenbank 61 67
62 Brenda Enzymdatenbank 62 68
63 Protein Data Bank 3D Strukturen 63 69
64 Biologische Datenbanken: Definition und Aufbau Basierend auf: Francis Ouellette Current Topics in Genome Analysis
65 Datenbanken Eine Datenbank ist eine systematisch strukturierte Sammlung von Informationen Daten, die man einmal darin gespeichert hat, können daraus wieder abgerufen werden. Durch Abfragen können neue Zusammenhänge zwischen den Daten gefunden werden. Eine Datenbank vereinfacht und reduziert den Datenraum durch Spezialisierung (Fokussierung) auf einen bestimmten Teilbereich. Eine Datenbank kann eine Ressource für andere Datenbanken und für Analysewerkzeuge sein
66 Datenbank Komponenten Definition und Beschreibung der Struktur Eindeutigen Schlüssel Links zu anderen Datenbanken Dokumentation Prozess für das Einreichen, Aktualisieren und Korrigieren von Einträgen 66 74
67 Datenbank Design.. The more closely and elegantly a model follows a real phenomenon, the more useful it is in predicting or understanding the natural phenomenon it mimics. Ostell J, et al. The NCBI Data Model. In: Bioinformatics, a Practical Guide to the Analysis of Genes and Proteins., Eds: Baxevanis AD and Ouellette BF. John Wiley & Sons, New York, 1998:p
68 Das NCBI Daten Model ist in ASN.1 definiert ASN.1 (Abstract Syntax Notation) ist eine Datenbeschreibungssprache ähnlich einer Backus-Naur Normalform. ASN.1 ist eine formale Sprache, die speziell dafür geschaffen wurde, komplexe Datenstrukturen in einer Form zu spezifizieren, die unabhängig von Computer, Betriebssystem, DBMS und Programmiersprache ist. ASN.1 ist ein internationaler Standard (ISO 8824, 8825) ASN.1 wird in vielen Protokollen zum Datenaustausch verwendet (e.g. X.400 or Z39.50)
69 Eine Bioseq definiert ein Koordinatensystem für Sequenzen ASN.1 Definition Bioseq ::= SEQUENCE { } id SET OF Seq-id, descr Seq-descr OPTIONAL, inst Seq-inst, annot SET OF Seq-annot OPTIONAL Die einfachste Bioseq besteht aus einer ID und der Sequenzinstanz (z.b. Länge, Topologie, Residuen)
70 Es gibt viele Klassen einer Bioseq Eine Bioseq kann eine DNA-, RNA-, oder Proteinsequenz beschreiben Eine Bioseq kann in verschiedenen Formen repräsentiert werden Eine Bioseq kann eine Historie (Seq-hist) haben 70 78
71 Seq-id s haben unterschiedliche Formen und Bedeutungen Eine Seq-id ist definiert als eine Menge von unterschiedlichen Typen, die unterschiedliche Formate und Bedeutungen haben. Einige davon spiegeln die Form und Anwendung in der Quelldatenbank wider. Die NCBI gi ist ein beliebiger ganzzahliger Identifier, der: explizit eine spezifische Sequenz identifiziert stabil ist und unverändert über die Zeit abgerufen werden kann die selbe Form über alle Sequenzdatenbanken hat es ermöglicht, eine Historie der Änderungen zu einer Sequenz zu schaffen 71 79
72 Zusätzliche Ressourcen Full dump der Daten (über FTP) Dokumentation der Datenbanken Programmatische Suche in den Datenbanken (Entrez Programming Utilities) Einheitliche Abfragesprache Dokumentation der Software für den Datenzugriff Details:
73 Primärdaten DNA-, RNA- und Proteinsequenzen sind Primärdaten in den biologischen Datenbanken. In den meisten Fällen sind die Proteinsequenzen von den DNA-, bzw. RNA-Sequenzen abgeleitet. Das Verständnis der unterschiedlichen Sequenztypen ist eine wesentliche Vorraussetzung für die Interpretation und Analyse der Sequenzen. Trotz aller Sorgfalt enthalten biologische Datenbanken Fehler. Das muss bei allen Abfragen berücksichtigt werden und die Resultate immer kritisch betrachtet werden
74 Was umfasst GenBank? GenBank ist die NIH Sequenzdatenbank aller öffentlich verfügbaren DNA- und deren abgeleiteten Protein Sequenzen. Jede dieser Sequenzen enthält Annotationen, die zusätzlichen biologische Informationen enthalten. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. Nucleic Acids Res. 2009;37(Database issue):d
75 Nucleotide Tägliche Synchronisation zwischen den INSDEC Datenbanken 75 83
76 GenBank - Release Oktober ,335,396 Einträge (Sequenzen oder GBFFs) 155,176,494,699 Nukleotide Vollständige Release von GenBank: alle 2 Monate. Inkrementelle und kumulative Releases: täglich
77 Einige Bemerkungen zu GenBank GenBank ist ein nukleotide-zentrischer Blick auf den Datenraum. GenBank ist ein Repository für alle öffentlich verfügbaren Sequenzen. Was nicht in GenBank gefunden werden kann, ist nicht in der public domain. GenBank Einträge sind nach unterschiedlichen Kriterien gruppiert. Das Verstehen dieser Gruppierungen ist essentiell für eine optimale Nutzung. Die Qualität der Daten in GenBank (und in biologischen Datenbanken i.a.) hängt primär vom Einreicher ab: Das muss sowohl bei Abfragen als auch beim Einreichen berücksichtigt werden
78 GBFF and ASN.1 GenBank Daten werden am NCBI im ASN.1 Format verwaltet. ASN.1 als Datenbeschreibungssprache ist i.a. nicht dazu gedacht von einem Menschen gelesen zu werden, sondern durch einen Computer interpretiert zu werden. Deshalb stellt GenBank unterschiedliche Ansichten auf die Daten. Das GenBank Flat File (GBFF) ist eine dieser Ansichten, das aus dem ASN.1 Format generiert werden kann
79 ASN.1 als lingua franca der Bioinformatik 79 87
80 Aufbau eines GenBank Flat Files (GBFFs) LOCUS HSU bp mrna linear PRI 21-MAY-1998 DEFINITION Homo sapiens integrin-linked kinase (ILK) mrna, complete cds. ACCESSION U40282 VERSION U GI: KEYWORDS. SOURCE human. ORGANISM Homo sapiens Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo. REFERENCE 1 (bases 1 to 1789) AUTHORS Hannigan,G.E., Leung-Hagesteijn,C., Fitz-Gibbon,L., Coppolino,M.G., Radeva,G., Filmus,J., Bell,J.C. and Dedhar,S. TITLE Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase JOURNAL Nature 379 (6560), (1996) MEDLINE REFERENCE 2 (bases 1 to 1789) AUTHORS Dedhar,S. and Hannigan,G.E. TITLE Direct Submission JOURNAL Submitted (07-NOV-1995) Shoukat Dedhar, Cancer Biology Research, Sunnybrook Health Science Centre and University of Toronto, 2075 Bayview Avenue, North York, Ont. M4N 3M5, Canada REFERENCE 3 (bases 1 to 1789) AUTHORS Dedhar,S. and Hannigan,G.E. TITLE Direct Submission JOURNAL Submitted (21-MAY-1998) Shoukat Dedhar, Cancer Biology Research, Sunnybrook Health Science Centre and University of Toronto, 2075 Bayview Avenue, North York, Ont. M4N 3M5, Canada REMARK Sequence update by submitter COMMENT On May 21, 1998 this sequence version replaced gi: FEATURES Location/Qualifiers source /organism="homo sapiens" /db_xref="taxon:9606" /chromosome="11" /map="11p15" /cell_line="hela" gene /gene="ilk" CDS /gene="ilk" /note="protein serine/threonine kinase" /codon_start=1 /product="integrin-linked kinase" /protein_id="aac " /db_xref="gi: " /translation="mddiftqcregnavavrlwldntendlnqgddhgfsplhwacre GRSAVVEMLIMRGARINVMNRGDDTPLHLAASHGHRDIVQKLLQYKADINAVNEHGNV PLHYACFWGQDQVAEDLVANGALVSICNKYGEMPVDKAKAPLRELLRERAEKMGQNLN RIPYKDTFWKGTTRTRPRNGTLNKHSGIDFKQLNFLTKLNENHSGELWKGRWQGNDIV VKVLKVRDWSTRKSRDFNEECPRLRIFSHPNVLPVLGACQSPPAPHPTLITHWMPYGS LYNVLHEGTNFVVDQSQAVKFALDMARGMAFLHTLEPLIPRHALNSRSVMIDEDMTAR ISMADVKFSFQCPGRMYAPAWVAPEALQKKPEDTNRRSADMWSFAVLLWELVTREVPF ADLSNMEIGMKVALEGLRPTIPPGISPHVCKLMKICMNEDPAKRPKFDMIVPILEKMQ DK" BASE COUNT 443 a 488 c 480 g 378 t ORIGIN 1 gaattcatct gtcgactgct accacgggag ttccccggag aaggatcctg cagcccgagt 61 cccgaggata aagcttgggg ttcatcctcc ttccctggat cactccacag tcctcaggct 121 tccccaatcc aggggactcg gcgccgggac gctgctatgg acgacatttt cactcagtgc 181 cgggagggca acgcagtcgc cgttcgcctg tggctggaca acacggagaa cgacctcaac 241 cagggggacg atcatggctt ctcccccttg cactgggcct gccgagaggg ccgctctgct 301 gtggttgaga tgttgatcat gcggggggca cggatcaatg taatgaaccg tggggatgac 361 acccccctgc atctggcagc cagtcatgga caccgtgata ttgtacagaa gctattgcag 421 tacaaggcag acatcaatgc agtgaatgaa cacgggaatg tgcccctgca ctatgcctgt 481 ttttggggcc aagatcaagt ggcagaggac ctggtggcaa atggggccct tgtcagcatc 541 tgtaacaagt atggagagat gcctgtggac aaagccaagg cacccctgag agagcttctc 601 cgagagcggg cagagaagat gggccagaat ctcaaccgta ttccatacaa ggacacattc 661 tggaagggga ccacccgcac tcggccccga aatggaaccc tgaacaaaca ctctggcatt 721 gacttcaaac agcttaactt cctgacgaag ctcaacgaga atcactctgg agagctatgg 781 aagggccgct ggcagggcaa tgacattgtc gtgaaggtgc tgaaggttcg agactggagt 841 acaaggaaga gcagggactt caatgaagag tgtccccggc tcaggatttt ctcgcatcca 901 aatgtgctcc cagtgctagg tgcctgccag tctccacctg ctcctcatcc tactctcatc 961 acacactgga tgccgtatgg atccctctac aatgtactac atgaaggcac caatttcgtc 1021 gtggaccaga gccaggctgt gaagtttgct ttggacatgg caaggggcat ggccttccta 1081 cacacactag agcccctcat cccacgacat gcactcaata gccgtagtgt aatgattgat 1141 gaggacatga ctgcccgaat tagcatggct gatgtcaagt tctctttcca atgtcctggt 1201 cgcatgtatg cacctgcctg ggtagccccc gaagctctgc agaagaagcc tgaagacaca 1261 aacagacgct cagcagacat gtggagtttt gcagtgcttc tgtgggaact ggtgacacgg 1321 gaggtaccct ttgctgacct ctccaatatg gagattggaa tgaaggtggc attggaaggc 1381 cttcggccta ccatcccacc aggtatttcc cctcatgtgt gtaagctcat gaagatctgc 1441 atgaatgaag accctgcaaa gcgacccaaa tttgacatga ttgtgcctat ccttgagaag 1501 atgcaggaca agtaggactg gaaggtcctt gcctgaactc cagaggtgtc gggacatggt 1561 tgggggaatg cacctcccca aagcagcagg cctctggttg cctcccccgc ctccagtcat 1621 ggtactaccc cagcctgggg tccatcccct tcccccatcc ctaccactgt gcgcaagagg 1681 ggcgggctca gagctttgtc acttgccaca tggtgtcttc caacatggga gggatcagcc 1741 ccgcctgtca caataaagtt tattatgaaa aaaaaaaaaa aaaaaaaaa // Header Features Sequence 80 88
81 Beispiel eines GenBank Eintrags LOCUS HSU bp mrna linear PRI 21-MAY-1998 DEFINITION Homo sapiens integrin-linked kinase (ILK) mrna, complete cds. ACCESSION U40282 VERSION U GI: KEYWORDS. SOURCE human. ORGANISM Homo sapiens Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo. REFERENCE 1 (bases 1 to 1789) AUTHORS Hannigan,G.E., Leung-Hagesteijn,C., Fitz-Gibbon,L., Coppolino,M.G., Radeva,G., Filmus,J., Bell,J.C. and Dedhar,S. TITLE Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase JOURNAL Nature 379 (6560), (1996) MEDLINE REFERENCE 2 (bases 1 to 1789) AUTHORS Dedhar,S. and Hannigan,G.E. TITLE Direct Submission JOURNAL Submitted (07-NOV-1995) Shoukat Dedhar, Cancer Biology Research, Sunnybrook Health Science Centre and University of Toronto, 2075 Bayview Avenue, North York, Ont. M4N 3M5, Canada 81 89
82 Beispiel eines GenBank Eintrags FEATURES Location/Qualifiers source /organism="homo sapiens" /db_xref="taxon:9606" /chromosome="11" /map="11p15" /cell_line="hela" gene /gene="ilk" CDS /gene="ilk" /note="protein serine/threonine kinase" /codon_start=1 /product="integrin-linked kinase" /protein_id="aac " /db_xref="gi: " /translation="mddiftqcregnavavrlwldntendlnqgddhgfsplhwacre GRSAVVEMLIMRGARINVMNRGDDTPLHLAASHGHRDIVQKLLQYKADINAVNEHGNV PLHYACFWGQDQVAEDLVANGALVSICNKYGEMPVDKAKAPLRELLRERAEKMGQNLN RIPYKDTFWKGTTRTRPRNGTLNKHSGIDFKQLNFLTKLNENHSGELWKGRWQGNDIV VKVLKVRDWSTRKSRDFNEECPRLRIFSHPNVLPVLGACQSPPAPHPTLITHWMPYGS LYNVLHEGTNFVVDQSQAVKFALDMARGMAFLHTLEPLIPRHALNSRSVMIDEDMTAR ISMADVKFSFQCPGRMYAPAWVAPEALQKKPEDTNRRSADMWSFAVLLWELVTREVPF ADLSNMEIGMKVALEGLRPTIPPGISPHVCKLMKICMNEDPAKRPKFDMIVPILEKMQ DK" BASE COUNT 443 a 488 c 480 g 378 t ORIGIN 1 gaattcatct gtcgactgct accacgggag ttccccggag aaggatcctg cagcccgagt 61 cccgaggata aagcttgg ccgcctgtca caataaagtt tattatgaaa aaaaaaaaaa aaaaaaaaa 82 90
83 FASTA - Format 1. Zeile beginnt mit >, gefolgt von der Bezeichnung der Sequenz (keine Leerzeichen!), Text nach dem ersten Leerzeichen ist Kommentar und wird von den Programmen meist ignoriert >gi gb M CAJRR16SC Campylobacter fetus veneralis 16S TATGGAGAGTTTGATCCTGGCTCAGAGTGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAACGGAGT ATTAAGAGAGCTTGCNNNNTTAATACTTAGTGGCGCACGGGTGAGTAATGTATAGTTAATCTGCCCTACA CTGGAGGACAACAGTTAGAAATGACTGCTAATACTCCATACTCCTTCTTAACATAAGTTAAGTCGGGAAA GTNTTTCGGTGTAGGATGAGACTATATTGTATCAGCTAGTTGGTAAGGTAATGGCTTACCAAGGCTNTGA CGCATAACTGGTCTGAGAGGATGATCAGTCACACTGGAACTGAGACACGGTCCAGACTCCNNCGGGAGGC AGCAGTAGGGAATATTGCTCAATGGGGGAAACCCTGAAGCAGCAACGCCGCGTGGAGGATGACACTNTTC GGAGCGTAAACTCCNTTTGTTAGGGAAGAACCATGACGGTACCTAACGAATAAGCACCGGCTAACTCCGT GCCAGCAGCCGCGGTNATACGGAGGGTGCNAGCGTTACTCGGAATCACTGGGCGTAAAGGACGCGTAGGC GGATTATCAAGTCTTTTGTGAAATCTAACAGCTTAACTGTTAAACTGCTTGAGAAACTGATAATCTAGAG TGAGGGAGAGGCAGATGGAATTGGTGGTGTAGGGGTAAAATCCGTAGAGATCACCAGGAATACCCATTGC GAAGGCGATCTGCTGGAACTCAACTGACGCTAATGCGTGAAAGCGTGGGGAGCAAACAGGATTNGATACC Nucleotide Sequenz 1 Buchstabencode 83 91
84 1- Buchstabencode für Nukleinsäuren Code description A C G T U Adenine Cytosine Guanine Thymine Uracil R Purine (A or G) Y Pyrimidine (C, T, or U) M K W S C or A T, U, or G T, U, or A C or G B C, T, U, or G (not A) D A, T, U, or G (not C) H A, T, U, or C (not G) V A, C, or G (not T, not U) N Any base (A, C, G, T, or U) 84 92
85 FASTA - Format >seq1 MEKTMSAILLVLYIFVLHLQYSEVHSLATTSDHDFSYLSFAYDATDLELEGSYDYVIVG >seq2 GGTSGCPLAATLSEKYKVLVLERGSLPTAYPNVLTADGFVYNLQQEDDGKTPVERFVSED >seq3 GIDNVRGRVLGGTSIINAGVYARANTSIYSASGVDWDMDLVNQTYEWVEDTIVYKPNSQS Im FASTA Format können mehrere Sequenzen hintereinander in derselben Datei abgelegt werden und an Analyse Programme übergeben werden (z.b. multiples Sequenzalignment) 85 93
86 1- und 3-Buchstabencode für Aminosäuren Aminosäure 3-Buchst. 1-Buchst. Alanin Ala A Arginin Arg R Asparagin Asn N Asparaginsäure Asp D Cystein Cys C Glutamin Gln Q Glutaminsäure Glu E Glycin Gly G Histidin His H Isoleucin Ile I Leucin Leu L Lysin Lys K Methionin Met M Phenylalanin Phe F Prolin Pro P Serin Ser S Threonin Thr T Tryptophan Trp W Tyrosin Tyr Y Valin Val V zusätzliche Codes: Asp/Asn Asx B Glu/Gln Glx Z Selenocystein Sec U Pyrrolysin Pyl O 86 94
87 LOCUS, Accession, Accession.version & gi LOCUS: HSU40282 Nucleotide GI: ACCESSION: U VERSION: U Protein GI: ACCESSION: AAC16892 VERSION: AAC
88 LOCUS, Accession, Accession.version & gi LOCUS: eindeutige ID, bestehend aus 10 Buchstaben und Ziffern. Wird nicht datenbankübergreifend gewartet und ist daher eine schlechte Sequenz-ID. ACCESSION: eine eindeutige ID, die sich nicht ändert, wenn ein Eintrag aktualisiert wird. Eine gute Sequenz-ID, die in Publikationen verwendet werden soll Nucleotide gi: Geninfo identifier (gi), eine eindeutige, ganzzahlige Sequenz-ID, die sich jedes Mal ändert, wenn sich die Sequenz ändert. Accession.version: eindeutige ID für einen Eintrag. Die Version wird inkrementiert, wenn die Sequenz sich ändert. Protein gi: gleich wie Nucleotide gi, für die Proteinsequenz protein_id: gleich wie Nucleotide Accession.version 88 96
89 GenBank Release 186 nach Organismen Entries Bases Species Homo sapiens Mus musculus Rattus norvegicus Bos taurus Zea mays Sus scrofa Danio rerio Strongylocentrotus purpuratus Oryza sativa Japonica Group Nicotiana tabacum Xenopus (Silurana) tropicalis Arabidopsis thaliana Drosophila melanogaster Pan troglodytes Vitis vinifera Canis lupus familiaris Gallus gallus Glycine max Macaca mulatta Solanum lycopersicum 89 97
90 GenBank Organismal divisions PRI -Primate ROD - Rodent MAM - Mammalian VRT - Vertebrate INV - Invertebrate PLN - Plant, fungal, and algal BCT -Bacterial VRL -Viral PHG - Phage SYN - Synthetic UNA - Unannotated 90 98
91 GenBank Functional Divisions PAT -Patent EST - Expressed Sequence Tags STS - Sequence Tagged Sites GSS - Genome Survey Sequences HTG - High Throughput Genome HTC - unfinished high-throughput cdna sequencing ENV - environmental sampling sequences 91 99
92 EST: Expressed Sequence Tag Expressed Sequence Tags sind einzelne, kurze ( bp) Reads von mrna (cdna), die in einem Hochdurchsatzverfahren generiert werden. Sie repräsentieren eine Momentaufnahme der Gene, die in einem gewissen Gewebe zu einem gewissen Zeitpunkt (z.b. Entwicklungszustand) expremiert (aktiv) sind
93 GSS: Genome Survey Sequences Genome Survey Sequences sind ähnlich zu ESTs, mit dem Unterschied, das sie Abschnitte genomischer DNA representieren und nicht cdna (reverse transcribed mrna)
94 Download Formate GBFF GenBank GBFF GenBank (vollständig) FASTA Nur Header und Sequenz FASTA Feature Tabelle ASN.1 GenBank vollständig XML GenBank (vollständig) XML International Nucleotide Sequence Database Collaboration content XML Tiny sequence
95 Prinzipien in der GenBank In GenBank werden Einträge nach verschiedenen Kategorien gruppiert, das betrifft sowohl organismusbezogene als auch funktionsbezogene Gruppen Das Verständnis dafür ist für die vollständige Nutzung dieser Datenbank essentiell
96 Bioinformatik: Next Generation Sequencing: Technologien Gerhard Thallinger Institut für Genomik und Bioinformatik, TU Graz
97 Entwicklung der Sequenziertechnologie First Generation 1977 Sanger Sequenzierung (GOLD Standard) Second Generation Sequencing (von Roche aufgekauft) 2005 Solexa Sequencing (von Ilumina aufgekauft) 2006 APG SOLiD Sequencing (von Life Technologies aufgekauft) Third Generation Helicos 2008 Pacific Biosciences 2011 Ion Torrent (von Life Technologies aufgekauft) Nanopore Sequencing (Roche & IBM)
98 Entwicklung der Sequenzierkosten
99 Sequenzierung eines Humanen Genoms 2001: Human Genome Project 2.7G$, 11 Jahre Log 10 (Kosten) : Celera 100M$, 3 Jahre 2007: 454 1M$, 3 Monate : SOLiD / Life 60K$, 2 Wochen 2009: Illumina, Helicos 40-50k$ 2010: Complete Genomics. 5k$, 1 W. 2014: 100$, <24 h? 2010 Jahr
100 Begriffe Library Pool von DNA Fragmenten, der aus einer Probe nach einem bestimmten Protokoll generiert wurde Run Sequenzierlauf mit einer oder mehreren Libraries Read Eine DNA-Sequenz, die in einem Run generiert wird (eine von sehr vielen, siehe Ausbeute) Read Länge Länge eines Reads (in basepairs [bp] angegeben) Ausbeute Gesamtanzahl der bp, die in einem Run generiert wird (in Gbp) errechnet sich aus Anzahl der Reads * der durchschnittlichen Read Länge
101 Zweite Generation: 454 / Roche Library Generierung mit emulsion PCR Erzeugung der Reads über pyrosequencing Read Länge ~750bp Ausbeute ~1 Gbp An sich geringe Fehlerrate, aber Probleme bei Homopolymer Abschnitten: z.b.: AAAAAAAAAA -> eff. Fehlerrate ~1% Margulies M, et al., Nature. 2005;437(7057): Mardis ER. Annu Rev Genomics Hum Genet. 2008;9:
102 Zweite Generation: 454 Pyrosequencing Während der DNA Synthese wird ein dntp an das 3 Ende des DNA Stranges angehängt. Die beiden Phosphatgruppen am Ende des dntp werden als Pyrophosphat (PPi) freigegeben. ATP sulfurylase erzeugt über PPi und Adenosine 5 -phosphosulfat ATP. Das Enzym Luciferase erzeugt das Leuchten der Glühwürmchen. Luciferin und ATP sind die Substrate, es werden dabei Luciferin zu Oxyluciferin unter der Abgabe von sichtbaren Licht konvertiert. Nach Ende der Reaktion wird Apyrase hinzugefügt, um die überschüssigen dntps zu zerstören
103 Zweite Generation: Solexa / Illumina Library Generierung mit bridge PCR Erzeugung der Reads mit Polymerase und reversible terminators Read Länge ~50-250bps Ausbeute ~600 Gbps Etwas höhere Fehlerrate als 454 Bentley DR, et al. Nature. 2008;456(7218): Mardis ER. Annu Rev Genomics Hum Genet. 2008;9:
104 Zweite Generation: Solexa / Illumina
105 Zweite Generation: Solid / Life Technologies Library Generierung wie bei 454 Read Länge ~100 bp Ausbeute ~100 Gbp Geringe Fehlerrate Complementary strand elongation mit Ligase und speziellen 8-mer probes Jede Position wird 5-mal gelesen Ausgabe der Sequenz im Colorspace, daher sehr empfindlich auf Fehler einer einzelnen Base die gesamte nachfolgende Sequenz ändert sich Shendure J, et al., Science 309 (2005) Mardis ER. Annu Rev Genomics Hum Genet. 2008;9:
106 Zweite Generation: SOLiD / Life Technologies
107 Zweite Generation: SOLiD / Life Technologies
108 Dritte Generation: Helicos Single molecule sequencing, keine Amplifizierung notwendig Ligation eines poly-a zur Fixierung auf einem Array Read Länge: ~ 50bp Ausbeute: 25 Gbp Lipson D, et al., Nat Biotechnol. 2009;27(7):
109 Dritte Generation: Pacific Biosciences Single molecule sequencing, keine Amplifizierung notwendig Realtime sequencing, Basenabfolge kann direkt abgelesen werden Die Geschwindigkeit der Polymerase kann gemessen werden Methylierung einer Base kann detektiert werden Eid J, et al. Science. 2009;323(5910):
110 Dritte Generation: Pacific Biosciences Circular Sequencing, die DNA Sequenz wird mehrfach gelesen um eine höhere Genauigkeit zu erreichen Read Länge: bis zu 25kps, Ø 3kbp, Ausbeute 0,2 Gbp, 15% Fehler (Insertionen und Deletionen), Laufzeit: ~1h
111 Dritte Generation: Pacific Biosciences
112 Dritte Generation: Ion Torrent ph sensing Library Generierung wie bei Roche 454 Read Länge: ~100bp, Ausbeute 1 Gbp, Laufzeit: ~2h ( 318 chip) Fehlerrate: ~1% (Homopolymers) Rothberg JM, et al. Nature 2011;475(7356):
113 Dritte Generation: Ion Torrent / Life Techn Ablauf wie bei 454 in Zyklen: Nukleotide werden in definierter Abfolge eingebracht und die ph Wert Änderung pro Well gemessen Welldichte (Anzahl Wells pro Chip) definiert die Ausbeute
114 Vierte Generation: Nanopore DNA Strang läuft durch eine Pore Die Spannungsänderung pro Nukleotid / Nukleotidpaar wird gemessen Anzahl der Poren definiert die Ausbeute Readlänge & Ausbeute noch unbekannt Fehlerrate noch unbekannt Clarke J, et al. Nat Nanotechnol. 2009;4(4):
115 Eckdaten der Technologien Hersteller Sanger / ABI Generierung der Library PCR, Clonal amplification 454 / Roche Emulsion PCR Solexa / Ilumina Bridge PCR Generierung der Reads Polymerase (Dideoxy sequen.) Polymerase (pyrosequencing) Polymerase (reversible termin.) Kosten pro Mb ($) Gerätekosten (k$) Read Länge (bp) Ausbeute (Gbp) Fehler rate (%) Lauf -zeit (d) ~5000 ~ , ,01 ~ ,5 ~ bis zu > SOLiD / Life Technologies Helicos Pacific Biosciences Ion Torrent / Life Techn. Emulsion PCR Single Molecule Single Molecule Emulsion PCR Ligase (octamers 2 base encoding) Polymerase (asyn. extensions) ~ bis zu 100 ~ bis zu 55 Polymerase ~1 625 bis zu Polymerase (ph sensing) 155 > ~25 ~4 2 0,2 ~15 0,05 ~1 50/200 > Adapted from Glenn TC. Molecular Ecology Resources 2011;11(5):
116 Next Generation Sequencing Details zur Biochemie unter
117 Bioinformatik: Next Generation Sequencing: Sequenz Assembly und Annotation Gerhard Thallinger Institut für Genomik und Bioinformatik, TU Graz
118 NGS Anwendungen Genom Sequenzierung Genom Re-Sequenzierung Transkriptom Sequenzierung (EST Sequenzierung) SNP Analyse Gen-Expressionsanalyse (RNA-seq) microrna Profiling Bestimmung der Transcription start site (TSS) Analyse der Transkriptionsdynamik (global run-on sequencing, GROseq) Transkriptionsfaktor Analyse (ChIP-seq) Globale Enhancer Aktivität (STARR-seq) Nukleosome Positionierung Methylierungsanalyse Microbiome Analyse Metagenome Analyse
119 Genom Sequenzierung: Ablauf Sequenzierung -> Rohdaten De novo Assemblierung -> Contigs / Scaffolds Finishing -> Genomsequenz (1 oder mehrere Chromosomen, Plasmide, Mitochondrium) Annotation -> annotiertes Genom (GBFF oder EMBL File) Einreichen (EMBL Nucleotide, GenBank oder DDBJ)
120 Anwendung: Genome Sequenzierung TG..GT TC..CC AC..GC CG..CA TT..TC AC..GC GA..GC TG..AC CT..TG GT..GC AC..GC AC..GC AA..GC AT..AT TT..CC Genome Kurze DNA Fragmente Kurze DNA Sequenzen de novo Assembly ACGTGGTAA CGTATACAC TAGGCCATA GTAATGGCG CACCCTTAG TGGCGTATA CATA ACGTGGTAATGGCGTATACACCCTTAGGCCATA ACGTGACCGGTACTGGTAACGTACA CCTACGTGACCGGTACTGGTAACGT ACGCCTACGTGACCGGTACTGGTAA CGTATACACGTGACCGGTACTGGTA ACGTACACCTACGTGACCGGTACTG GTAACGTACGCCTACGTGACCGGTA CTGGTAACGTATACCTCT... Genomsequenz
121 Anwendung: Genome Sequenzierung Contig: Ein contig ist eine zusammenhängender Abschnitt einer genomischen Sequenz, die aus überlappenden Reads gebildet wird. Paired-end (mate-pair) reads: Read Paar mit einem definierten Abstand auf dem Genom Scaffold: Ein Scaffold besteht aus Contigs und Gaps. Die Gap Länge kann aus paired-end bzw. matepair Information abgeschätzt werden. Green 2001, Nature Reviews Genetics 2:
122 Genome Assemblierung Relativ einfach in einer perfekten Welt Ohne Sequenzierfehler Ohne Repeats Mit uniformer Abdeckung (Coverage) Assemblierung mit O(n²) Aufwand (n=anzahl der Reads) In der Realität Chimerische Sequenzen aufgrund der PCR Verschiedenartige Sequenzierfehler (Fehler in der Länge von Homopolymerabschnitten, Indels, fehlerhafte Basen) Repeats unterschiedlicher Länge Genomeabschnitte ohne (mit geringer) Abdeckung
123 Genome Assemblierung Grundprinzip: Kombiniere alle Reads basierend auf Sequenzähnlichkeit in Es wird angenommen, dass überlappende Reads aus dem selben Bereich im Genom kommen Contigs müssen nachbearbeitet werden (Finishing): Assembler erzeugen i.a. zu viele Contigs. Finishing nimmt die Contigs als Basis um eine vollständige Genomsequenz zu bilden. Scaffolder reihen Contigs in Scaffolds basierend auf der paired-end / mate-pair Information. Jeder Scaffold enthält Gaps, die sich aufgrund der Reihenfolge der Contigs ergeben. Sequenzgaps sind Gaps zwischen Contigs im selben Scaffold. Physische Gaps sind Gaps zwischen Scaffolds (entstehen aufgrund ungenügender / fehlender paired-end / mate-pair Sequenzen)
124 Assemblierung: Algorithmen Contig Bildung: Greedy Algorithmus Finde den kürzesten gemeinsamen Superstring einer Menge an Read Ausgehend von den Reads {s 1, s 2, } finde den kürzesten String T, so dass jeder Read s i ein Teilstring von T ist. Die Lösung dieses Problems ist sehr zeitaufwändig (NP-hard) aber kann durch einen Greedy Algorithmus effizient angenähert werden. Greedy Algorithmen wurden erfolgreich für das Assembly von Bakterien und einzelligen Eukaryonten eingesetzt Limitierung: realtiv langsam für grosse Anzahl von Reads Verwendet von phrap, TIGR, CAP3 für Sanger-Daten, SSAKE, VCAKE, SHARCGS für NGS-Daten
125 Assemblierung: Algorithmen Contig Bildung: Overlap-layout-consensus Der Algorithmus basiert auf Graphentheorie Es wird ein Graph konstruiert dessen Knoten den Reads entsprechen Kanten überlappende Reads entsprechen Ein Contig ist dann ein einfacher Pfad im Graphen Ein einfacher Pfad ist ein Pfad durch den Graphen, der einen Knoten maximal einmal enthält Der Assembler konstruiert den Graphen Output ist eine Menge von sich nicht schneidenden einfachen Pfaden, wobei jeder Pfad einem Contig entspricht. Verwendet von Arachne, Celera Assembler, newbler, Minimus, Edena
126 Assemblierung: Algorithmen Contig Bildung: Eulersche Pfade / De Bruijn graph Der Algorithmus basiert ebenfalls auf Graphentheorie Eulscher Pfad: Ein Pfad, der alle Kanten eines Graphen besucht Reads werden in überlappende k-mers aufgespalten. Der De Bruijn Graph eines of eines Assemblies hat als Knoten alle eindeutigen k 1 mers Eine Kante wird dann gebildet, wenn es eine Überlappung zwischen dem k-1 Prefix eines k-mers und dem k-1 Suffix des k- mers gibt Das zugrundeliegende Problem wird nun reduziert auf das Finden eines Pfades, der alle Kanten im Graphen besucht. Der Ansatz mit Eulerschen Pfaden ist effizienter, in der Praxis sind die beiden Ansätze aber ungefähr gleich schnell. Verwendet von Euler, Velvet, Allpath, ABySS
127 Assemblierung: Algorithmen Contig Bildung: Eulersche Pfade / De Bruijn graph
128 Assemblierung: Repeat Behandlung Assembler sollten Repeat schon während des Assemblierungsprozesses erkennen Damit wird eine falsche Rekonstruktion des Genoms vermieden Assembler sollten so viele Repeats wie möglich auflösen. Damit können arbeitsintensive Nacharbeiten im Labor vermieden werden (PCR, weitere Sequenzierungsläufe)
129 Assemblierung: Repeat Behandlung Repeat Erkennung Annahme: Reads sind gleichförmig und zufällig verteilt Basierend auf dieser Annahme, können Repeats als Bereiche mit wesentlich höherer Coverage identifiziert werden. Leider ist diese Annahme of nicht erfüllt (abhängig von der Sequenziertechnologie, dem GC-Gehalt des Genoms, ) Repeats können auch über komplexen Teilgraphen des De Bruijn graph erkannt werden Paired-end bzw. mate-pair Information kann ebenfalls zur Repeat Identifikation verwendet werden (z.b. über Read-Paare die einen zu großen oder zu geringen Abstand haben) Geringe Unterschiede in der Repeatsequenz können zur korrekten Einbindung in die Genomsequenz verwendet werden
130 Assemblierung: Scaffolding Scaffolding ist der Prozess Contigs in Untermengen mit bekannter Reihenfolge und Orientierung zu bringen. Knoten sind Contigs Eine gerichtete Kante zwischen 2 Contigs wird dann eingeführt, wenn ein Read-Paar den dazwischenliegenden Gap überbrückt
131 Assemblierung: Scaffolding Probleme beim Scaffolding: Alle verbundenen Komponenten zu finden Eine konsistente Orientierung für alle Contigs im Graphen zu finden. Fehlerhafte Read-Paare (spurious links zwischen Contigs) Große Streuung bei der Insert-Länge (Abstand des Read-Paars) Genomsequenzen von engverwandten Species können für das Scaffolding verwendet werden (Achtung: Rearrangements) Programme: Bambus, SSPACE Newbler, Mira, Velvet (im Assembler integriert)
132 Genom Sequenzierung: Finishing Scaffold Reihenfolge / Orientierung: PCR Primer Design für die Enden aller Scaffolds (forward & reverse) PCR aller Primer Kombinationen PCR Reaktionen mit Amplikon -> Scaffolds an die die Primer binden sind benachbart Scaffold Reihenfolge / Orientierung: Optical Mapping DNA mit verschiedenen Restriktionsenzymen verdauen Bestimmung der Fragmentlänge mit Hilfe eines optischen Verfahrens In silico Verdau der unterschiedlichen Genomvarianten und Vergleich der Längen
133 Genom Sequenzierung: Finishing Gap closing: PCR Primer Design für die Enden der benachbarten Contigs PCR -> Amplikon (weil benachbart im Scaffold) Sanger Sequenzierung der Amplikons und Integration der Sequenz in das Genom Gap closing: in silico gap closing Verwendung der paired-end Reads Contigs in die Sequenz integrieren
134 Assemblierung: Abschätzung der Qualität Anzahl und Größe der Contigs Annahme: wenige große Contigs sind besser als viele kleine N50 Wert wird als Masszahl verwendet: Entspricht der Länge des kleinsten Contigs in einem Set der größten Contigs, deren zusammengerechnete Länge mindestens 50% des Assemblies beträgt Maximale und durchschnittliche Länge eines Contigs Anzahl der Scaffolds und Anzahl der Contigs im Scaffold Annahme: wenige Scaffold mit einer großen Anzahl von Contigs sind besser als viele mit wenigen Eine natürliche Untergrenze für die Anzahl der Scaffolds ist die Anzahl der Chromosomen S50 Wert wird als Masszahl verwendet (analog zu N50) Maximale und durchschnittliche Länge eines Scaffolds
135 Assemblierung: Abschätzung der Qualität Sequenzqualität Phred quality score (Q) ist das negative Verhältnis der Wahrscheinlichkeit einer fehlerhaften Base (P) bezogen auf die Referenz in Dezibel (db): Q = -10 log 10 P Relative Anzahl der fehlerhaften Basen Q30.. ~ eine fehlerhafte Base in 1000 Q60.. ~ eine fehlerhafte Base in
136 Genom Sequenzierung: Annotation Von der Sequenz. zur Liste von annotierten Genen
137 Genom Sequenzierung: Annotation Identifikation von funktionellen Sequenzabschnitten Genen Protein kodierende Gene nicht protein kodierende Gene, z.b. ncrnas (rrna, trna, mirnas, ) Funktionszuweisung der Proteine Identifikation von Transcription Start Sites und Bindestellen für Transkriptionsfaktoren
138 Gen Identifizierung: Terminologie Gen: "a locatable region of genomic sequence, corresponding to a unit of inheritance, which is associated with regulatory regions, transcribed regions, and or other functional sequence regions ". Pearson H. Nature 2006; 441(7092): Operon: Gruppe von Genen, die gemeinsam transkribiert werden (in Prokaryonten) 138
139 Gen Identifizierung: Terminologie Eine Abfolge von DNA Nukleotiden, die ein Gen enthält, hat unterschiedliche Regionen: Exons kodierende Regionen Introns nicht kodierende Regionen innerhalb eines Gens Getrennt durch funktionelle Sites (boxes) Start- (S) und Stopkodons (T) Splice sites Acceptors (A) und Donors (D)
140 Gen Identifizierung: Probleme Es muss mit dem korrekten Reading Frame begonnen werden Ein Intron kann ein Exon mitten in einem Kodon unterbrechen Es gibt keine schnelle und eindeutige Regel für die Identifizierung der Donor- und Acceptor Spliceposition Signale sind sehr schwach
141 HMM Gene Finders: AUGUSTUS Basiert auf einem generalisiertem HMM Kann Zusatzinformationen (z.b. Lage einzelner Exons) integrieren Kann cdna Alignments (ESTs) zur Verbesserung des Ergebnisses verwenden Stanke M, et al. Bioinformatics. 2008;24(5): Stanke M & Waack S. Bioinformatics. 2003;19(2):ii215-ii
142 HMM Gene Finders: GlimmerHMM Basiert auf einem generalisierten HMM Zusätzlich werden Splice site Modelle verwendet, die aus GeneSplicer adaptiert wurden Nutzt einen Decision tree aus GlimmerM und interpolierte Markov Modelle für die kodierenden und nicht kodierenden Sequenzabschnitte. Majoros WH, et al. Bioinformatics. 2004;20(16):
143 Gene Finders: Vergleich Gene Finder Vergleich anhand P. aroginosa. LESB58 Prodigal liefert das beste Ergebnis bei diesem GC-reichen Genom Glimmer sagt längere Gene voraus Homologie-basierende Methoden (Rast) liefern schlechte Ergebnisse AMIGene findet neue Gene durch die Analyse der Codon usage Die beste Strategie ist die Verwendung einer Kombination von Gene Findern, gefolgt von einer evidenzbasierten manuellen Überarbeitung
144 Identifikation von ncrnas: rrnas In Prokaryonten: 5S rrna (~120nt), 16S rrna (~1500nt), 23S rrna (~2900nt) Die DNA Sequenz ist stark konserviert Identifikation über Homologie RNAmmer ( RDP (( NAST ( Infernal (
145 Identifikation von ncrnas: trnas Zwischen 1 und 6 verschiedene trnas für jede Aminosäure Sehr kurz (74 95nt) Die DNA Sequenz ist sehr schwach konserviert, die Sekundärstruktur sehr stark Identikation über Modelle der Sekundärstruktur. RNAmmer ( trnascan-se (
146 Funktionsannotation Zuweisung über Homologie Suche nach bereits annotierten Proteinen mit ähnlicher Sequenz Übernahme der Annotation Funktion (Bezeichnung) Gene Ontologie (GO) Annotation Clusters of Orthologous Genes (COG) Zuordnung Enzymklasse Zuordnung zum Stoffwechselpfad
147 Funktionsannotation: Web Resources BaSys ( RAST ( IMG/ER ( JCVI (
148 Bioinformatik: Prädiktive Methoden basierend auf DNA und Protein Sequenzen Gerhard Thallinger Institut für Genomik und Bioinformatik, TU Graz
149 Umsetzung der genetischen Information Gen Funktion
150 Prädiktive Methoden Problem: Neue DNA oder Proteinsequenz, aber keine Information über die Funktion Idee: Vorhersage der Funktion über die Ähnlichkeit der Sequenz zu bereits bekannten, annotierten Sequenzen Neues Problem: Wie finde ich Sequenzen, die zur unbekannten Sequenz ähnlich sind
151 Protein Sequenz Analyse Protein Sequence Comparative Methods Predictive Methods Homology Profile Physical Structural Searches Analysis Properties Properties Gemeinsame Vorgänger (Vorfahren)? Ähnliche Funktion? Homologie auf Domain oder Sequenzebene?
152 BLAST BLAST Basic Local Alignment Search Tool Suche nach homologen Sequenzen durch Ausrichtung einer Sequenz zu einer anderen mit dem Ziel die größtmögliche Überdeckung zu erreichen Local bedeutet, dass Teile (Segmente) der Sequenz ausgerichtet werden und nicht die gesamte Sequenz (global) Erste Version von Altschul et al Weitere Version von Gish et al
153 BLAST Ablauf Sequenzen werden in Words gesplittet Seeds: identische Words in Query and Subject Suche nach high-scoring segment pairs (HSPs) Paare von Sequenzsegmenten die aligned (ausgerichtet) werden können Ausgehend von den Seeds wird das Alignment erweitert Die Segmentpaare haben nach dem Alignment den maximal erreichbaren Score (der Score kann durch Hinzufügen oder Weglassen von Teilen der Sequenz nicht mehr verbessert werden) Der Score muss über dem angegebenen Limit S liegen Zwei Versionen gapped (2.0) oder ungapped (1.4) Der Algorithmus beruht auf Dynamischer Programmierung mit einer Komplexität von O(n²), wobei n die Länge der längeren Sequenz ist
154 BLAST Ressourcen BLAST Web Service Kommandozeile blastall -p progname -d -db -i query > outfile Tutorial
155 Arten der BLAST Suche: DNA vs. DNA GCNTACACGTCACCATCTGTGCCACCACNCATGTCTCTAGTGATCCCTCATAAGTTCCAACAAAGTTTGC GCCTACACACCGCCAGTTGTG-TTCCTGCTATGTCTCTAGTGATCCCTGAAAAGTTCCAGCGTATTTTGC GAGTACTCAACACCAACATTGATGGGCAATGGAAAATAGCCTTCGCCATCACACCATTAAGGGTGA---- GAATACTCAACAGCAACATCAACGGGCAGCAGAAAATAGGCTTTGCCATCACTGCCATTAAGGATGTGGG TGTTGAGGAAAGCAGACATTGACCTCACCGAGAGGGCAGGCGAGCTCAGGTA TTGACAGTACACTCATAGTGTTGAGGAAAGCTGACGTTGACCTCACCAAGTGGGCAGGAGAACTCACTGA GGATGAGGTGGAGCATATGATCACCATCATACAGAACTCAC CAAGATTCCAGACTGGTTCTTG GGATGAGATGGAACGTGTGATGACCATTATGCAGAATCCATGCCAGTACAAGATCCCAGACTGGTTCTTG
156 Arten der BLAST Suche: Protein vs. Protein
157 DNA translated vs. Protein
158 DNA translated vs. DNA translated
159 BLAST Versionen Programm Query Sequenz Subject Sequenz BLASTN Nucleotide Nucleotide BLASTP Protein Protein BLASTX Nucleotide, six-frame translation Protein TBLASTN Protein Nucleotide, six-frame translation TBLASTX Nucleotide, six-frame translation Nucleotide, six-frame translation
160 BLAST Vorraussetzungen Eine query Sequenz, im FASTA Format Auswahl der BLAST Version Auswahl der Datenbank in der gesucht werden soll Festlegung der Suchparameter
161 BLAST: Aufbau der Suche paste your sequence here choose database specify search region nr ~ non-redundant database Others are subsets of nr database
162 BLAST: Optionen Beispiel: protease NOT hiv1[organism] Einschränkung der Suche auf einen Organismus Lower EXPECT thresholds are more stringent. The smaller the word size, the higher the sensitivity
163 BLAST: Filter Low-complexity: Einige Sequenzsegmente sind biologisch weniger interessant (z.b., Treffer in säure-, basen- oder Proline-reichen Regionen) die über die SEG bzw. DUST Programme identifiziert werden. Solche Segmente werden von der Suche ausgeschlossen. Human repeats: Mit dieser Option werden Repeats (LINE's und SINE's) im humanen Genom ausgeschlossen. Damit kann die Suchgeschwindigkeit wesentlich erhöht werden (besonders bei langen Sequenzen (>100 kb)). Mask for lookup table only: BLAST Suchen bestehen aus zwei Phasen: 1) Suche nach Hits mit Hilfe einer Tabelle und 2) Erweiterung der Hits. Mit dieser Option werden die ausgewählten Filter nur in der ersten Phase angewendet. Mask Lower Case: Sequenzen in Kleinbuchstaben werden nicht berücksichtigt. Damit können Filterregionen durch den Benutzer festgelegt werden
164 BLAST: Interpretation der Resultate query Sequenz BLAST Hits. Durch Klicken auf eine Zeile gelangt man zum paarweisen Alignment. Dieses Bild zeigt die Verteilung der BLAST Hits für die query Sequenz. Jede Zeile entspricht einem Hit. Der Bereich, den eine Linie überdeckt, repräsentiert die Region, wo eine Sequenzähnlichkeit gefunden wurde. Die verschiedenen Farben entsprechen Bereichen verschiedener BLAST- Scores (Bit scores)
165 BLAST: Interpretation der Resultate Die Beschreibungs- bzw. (Definitions-) Zeilen werden under der Überschrift "Sequences producing significant alignments angezeigt. "significant" bezieht sich auf alle Hits deren E-value geringer als das gewählte Limit ist. Das bedeutet jedoch keine biologische Signifikanz. Gene/Sequence Definition Expect value je kleiner desto besser. Representiert die Wahrscheinlichkeit eines zufälligen Hits ID (GI #, refseq #, DB-specific ID #) Über diesen Link gelangt man zur GenBank Bit score je höher desto besser. Über diesen Link gelangt man zum paarweisen Alignment Links
166 BLAST Resultat: Paarweises Alignment Query: Das Segment aus der query Sequenz. Subjct: Das Segment aus der hit (subject) Sequenz aus der Datenbank. Zwischenzeile: consensus Basen
167 BLAST Demo
168 Substitutions Matrizen Was ist eine Substitutionsmatrix? Eine 2-dimensionale Matrix mit den 4 Nukleotiden als Indices Werte in einer Zelle beinhalten den Score für den Austausch eines Nukleotids in der Query Sequenz um das Nukleotid an der selben Position in der Subject Sequenz zu erhalten Warum wird eine Substitutionsmatrix verwendet? Um die Wahrscheinlichkeit der Homologie zwischen zwei Sequenzen festzustellen. Substitutionen, die wahrscheinlicher sind, sollten einen höheren Score bekommen Substitutionen, die weniger wahrscheinlich sind, sollten einen geringeren Score bekommen
169 Substitutions Matrizen Wie sieht eine Substitutions-Matrix aus? A G C T A G C T
170 Beispiel anhand der Identity Matrix A G C T A G C T CAGGTAGCAAGCTTGCATGTCA raw score = 19 CACGTAGCAAGCTTG-GTGTCA
171 Beispiel anhand einer realistischeren Matrix A G C T A G C T CAGGTAGCAAGCTTGCATGTCA raw score = 19-9 = 10 CACGTAGCAAGCTTG-GTGTCA
172 Substitutions Matrizen Nukleotid Substitutions Matrizen, die bei Auswahl eines bestimmten Prozentsatzes der Sequenzähnlichkeit verwendet werden: % Sequenzidentität Match/Mismatch 99% 1/-3 98% 2/-5 95% 1/-2 90% 2/-3 85% 3/-4 80% 4/-5 75% 1/-1 70% 11/-10 65% 5/-4 60% 7/-5 50% 3/
173 Scoring Matrizen Matrix von Log-odds Werten, wo jede Zelle den Logarithmus des Wahrscheinlichkeitratios angibt, dass die betreffenden AA i durch die AA j substituiert bzw. aligned wird. Sij log beobachtete Anzahl Häufigkeit der AA von Substitutionen AAi nach AAj j Sie sind ein empirisches Schema zur Gewichtung eines Alignments mit Berücksichtigung der Biologie Cys/Pro sind wichtig für die Struktur und Funktion Trp hat große Seitenketten Lys/Arg haben positiv geladene Seitenketten
174 Scoring Matrizen Es ist wichtig, die Scoring Matrizen zu verstehen Sie werden in allen Analysen zum Sequenzvergleich verwendet Sie repräsentieren implizit eine gewissen Ansatz der Evolutionstheorie Die Wahl der Matrix beeinflusst das Resultat stark Score des Alignments = Summe der log-odds Werte aller AA Paare im Alignment
175 Struktur einer Scoring Matrix
176 PAM Matrizen Margaret Dayhoff, 1978 Point Accepted Mutation (PAM) Untersuchung der Substitutionsmuster in verwandten Proteinen (~1500 Proteine aus 71 Familien) Eine Mutation gilt als akzeptiert, wenn die Funktion der Seitenkette erhalten bleibt ( acceptance ) 1 PAM entspricht durchschnittlich der Änderung von einer Aminosäure pro 100 Residuen 1 PAM ~ 1% Sequenzunterschied Für Proteine mit größerem evolutionärem Abstand ist es notwendig, die Muster zu extrapolieren PAMx = PAM1 x Dayhoff MO, Schwartz RM, Orcutt BC. A Model of Evolutionary Change in Proteins. In: Atlas of Protein Sequence and Structure. National Biomedical Research Fundation. 1978;
177 PAM Matrizen Annahmen Die AA Substitution ist unabhängig von den benachbarten Residuen Die Wahrscheinlichkeit einer Mutation ist für alle Positionen in der Sequenz gleich Die Sequenzen, die verglichen wurden, repräsentieren die durchschnittliche Verteilung der AA Fehlerquellen Kleine, globuläre Proteine wurden zur Ableitung der Matrizen verwendet (Abweichung von der Annahme einer durchschnittlichen AA Verteilung) Fehler in PAM 1 werden vergrößern sich bis PAM 250 Konservierte Blöcke und Muster werden nicht berücksichtigt
178 BLOSUM Matrizen Henikoff und Henikoff, 1992 Blocks Substitution Matrix (BLOSUM) Es werden nur Unterschiede in konservierten Regionen (ohne Gaps) einer Proteinfamilie betrachtet. Sensitiver auf strukturelle oder funktionelle Substitutionen BLOSUM n Sequenzen mit > n% Identität werden mit 1 gewichtet Die Substitutionhäufigkeiten werden stärker von Sequenzen beeinflusst, die mehr als dieser Prozentsatz von einander abweichen Clustering reduziert den Beitrag von sehr ähnlichen Sequenzen Wahl einer Matrix mit kleinerem n liefert Sequenzen, die evolutionär weiter entfernt sind Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. PNAS. 1992;89(22):
Sequenziertechnologien
 Sequenziertechnologien Folien teilweise von G. Thallinger übernommen 11 Entwicklung der Sequenziertechnologie First Generation 1977 Sanger Sequenzierung (GOLD Standard) Second Generation 2005 454 Sequencing
Sequenziertechnologien Folien teilweise von G. Thallinger übernommen 11 Entwicklung der Sequenziertechnologie First Generation 1977 Sanger Sequenzierung (GOLD Standard) Second Generation 2005 454 Sequencing
Aufgabe 2: (Aminosäuren)
") Aufgabe 2: (Aminosäuren) Aufgabenstellung Die 20 Aminosäuren (voller Name, 1- und 3-Buchstaben-Code) sollen identifiziert und mit RasMol grafisch dargestellt werden. Dann sollen die AS sinnvoll nach ihren
Aufgabe 2: (Aminosäuren) Aufgabenstellung Die 20 Aminosäuren (voller Name, 1- und 3-Buchstaben-Code) sollen identifiziert und mit RasMol grafisch dargestellt werden. Dann sollen die AS sinnvoll nach ihren
Neue DNA Sequenzierungstechnologien im Überblick
 Neue DNA Sequenzierungstechnologien im Überblick Dr. Bernd Timmermann Next Generation Sequencing Core Facility Max Planck Institute for Molecular Genetics Berlin, Germany Max-Planck-Gesellschaft 80 Institute
Neue DNA Sequenzierungstechnologien im Überblick Dr. Bernd Timmermann Next Generation Sequencing Core Facility Max Planck Institute for Molecular Genetics Berlin, Germany Max-Planck-Gesellschaft 80 Institute
MOL.504 Analyse von DNA- und Proteinsequenzen. Datenbanken & Informationssysteme
 MOL.504 Analyse von DNA- und Proteinsequenzen Datenbanken & Informationssysteme Inhaltsübersicht Informationsysteme National Center for Biotechnology Information (NCBI) The European Bioinformatics Institute
MOL.504 Analyse von DNA- und Proteinsequenzen Datenbanken & Informationssysteme Inhaltsübersicht Informationsysteme National Center for Biotechnology Information (NCBI) The European Bioinformatics Institute
Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio
 Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio Ein Wissenschaftler erhält nach einer Sequenzierung folgenden Ausschnitt aus einer DNA-Sequenz: 5 ctaccatcaa tccggtaggt tttccggctg
Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio Ein Wissenschaftler erhält nach einer Sequenzierung folgenden Ausschnitt aus einer DNA-Sequenz: 5 ctaccatcaa tccggtaggt tttccggctg
MOL.504 Analyse von DNA- und Proteinsequenzen
 MOL.504 Analyse von DNA- und Proteinsequenzen Kurs 1 Monika Oberer, Karl Gruber MOL.504 Modul-Übersicht Einführung, Datenbanken BLAST-Suche, Sequenzalignment Proteinstrukturen Virtuelles Klonieren Abschlusstest
MOL.504 Analyse von DNA- und Proteinsequenzen Kurs 1 Monika Oberer, Karl Gruber MOL.504 Modul-Übersicht Einführung, Datenbanken BLAST-Suche, Sequenzalignment Proteinstrukturen Virtuelles Klonieren Abschlusstest
DATENQUALITÄT IN GENOMDATENBANKEN
 DATENQUALITÄT IN GENOMDATENBANKEN Alexander Fehr 28. Januar 2004 Gliederung Motivation Biologische Grundkonzepte Genomdaten Datenproduktion und Fehler Data Cleansing 2 Motivation (1) Genomdatenbanken enthalten
DATENQUALITÄT IN GENOMDATENBANKEN Alexander Fehr 28. Januar 2004 Gliederung Motivation Biologische Grundkonzepte Genomdaten Datenproduktion und Fehler Data Cleansing 2 Motivation (1) Genomdatenbanken enthalten
Foliensatz; Arbeitsblatt; Internet. Je nach chemischem Wissen können die Proteine noch detaillierter besprochen werden.
 03 Arbeitsauftrag Arbeitsauftrag Ziel: Anhand des Foliensatzes soll die Bildung und der Aufbau des Proteinhormons Insulin erklärt werden. Danach soll kurz erklärt werden, wie man künstlich Insulin herstellt.
03 Arbeitsauftrag Arbeitsauftrag Ziel: Anhand des Foliensatzes soll die Bildung und der Aufbau des Proteinhormons Insulin erklärt werden. Danach soll kurz erklärt werden, wie man künstlich Insulin herstellt.
ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCING DATA
 ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCING DATA Jason R. Miller*, Sergey Koren, Granger Sutton Ein Vortrag von Sergej Tschernyschkow Friedrich-Schiller-Universität Jena 03. Mai 2010 SERGEJ TSCHERNYSCHKOW
ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCING DATA Jason R. Miller*, Sergey Koren, Granger Sutton Ein Vortrag von Sergej Tschernyschkow Friedrich-Schiller-Universität Jena 03. Mai 2010 SERGEJ TSCHERNYSCHKOW
Homologie und Sequenzähnlichkeit. Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr
 Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und
 Welche besondere Art von Nukleotiden wird verwendet und") 10. Methoden 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und welche Funktion haben diese bei der Sequenzierungsreaktion?
10. Methoden 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und welche Funktion haben diese bei der Sequenzierungsreaktion?
MOL.504 Analyse von DNA- und Proteinsequenzen. Übungsaufgaben Datenbanken und Informationssysteme
 MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben Datenbanken und Informationssysteme Ü1 Tutorial für NCBI NCBI Nucleotide: Suche nach cellobiose dehydrogenase fungi Ü1 Tutorial für NCBI NCBI
MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben Datenbanken und Informationssysteme Ü1 Tutorial für NCBI NCBI Nucleotide: Suche nach cellobiose dehydrogenase fungi Ü1 Tutorial für NCBI NCBI
BIOINFORMATIK I ÜBUNGEN.
 BIOINFORMATIK I ÜBUNGEN http://icbi.at/bioinf Organisation 3 Übungen Kurze Einführung anschließend Labor Protokoll (je 2 Studierende, elektronisch doc, pdf..) Abgabe der Übungen bis spätestens 29. 05.
BIOINFORMATIK I ÜBUNGEN http://icbi.at/bioinf Organisation 3 Übungen Kurze Einführung anschließend Labor Protokoll (je 2 Studierende, elektronisch doc, pdf..) Abgabe der Übungen bis spätestens 29. 05.
DNA Sequenzierung. Transkriptionsstart bestimmen PCR
 10. Methoden DNA Sequenzierung Transkriptionsstart bestimmen PCR 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet
10. Methoden DNA Sequenzierung Transkriptionsstart bestimmen PCR 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet
MOL.504 Analyse von DNA- und Proteinsequenzen. Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
Primärstruktur. Wintersemester 2011/12. Peter Güntert
 Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Alignment-Verfahren zum Vergleich biologischer Sequenzen
 zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
Professionelle Seminare im Bereich MS-Office
 Der Name BEREICH.VERSCHIEBEN() ist etwas unglücklich gewählt. Man kann mit der Funktion Bereiche zwar verschieben, man kann Bereiche aber auch verkleinern oder vergrößern. Besser wäre es, die Funktion
Der Name BEREICH.VERSCHIEBEN() ist etwas unglücklich gewählt. Man kann mit der Funktion Bereiche zwar verschieben, man kann Bereiche aber auch verkleinern oder vergrößern. Besser wäre es, die Funktion
How to do? Projekte - Zeiterfassung
 How to do? Projekte - Zeiterfassung Stand: Version 4.0.1, 18.03.2009 1. EINLEITUNG...3 2. PROJEKTE UND STAMMDATEN...4 2.1 Projekte... 4 2.2 Projektmitarbeiter... 5 2.3 Tätigkeiten... 6 2.4 Unterprojekte...
How to do? Projekte - Zeiterfassung Stand: Version 4.0.1, 18.03.2009 1. EINLEITUNG...3 2. PROJEKTE UND STAMMDATEN...4 2.1 Projekte... 4 2.2 Projektmitarbeiter... 5 2.3 Tätigkeiten... 6 2.4 Unterprojekte...
Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2014
 Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2014 Fragen für die Übungsstunde 8 (14.07-18.07.) 1) Von der DNA-Sequenz zum Protein Sie können
Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2014 Fragen für die Übungsstunde 8 (14.07-18.07.) 1) Von der DNA-Sequenz zum Protein Sie können
Wie Google Webseiten bewertet. François Bry
 Wie Google Webseiten bewertet François Bry Heu6ge Vorlesung 1. Einleitung 2. Graphen und Matrizen 3. Erste Idee: Ranking als Eigenvektor 4. Fragen: Exisi6ert der Eigenvektor? Usw. 5. Zweite Idee: Die Google
Wie Google Webseiten bewertet François Bry Heu6ge Vorlesung 1. Einleitung 2. Graphen und Matrizen 3. Erste Idee: Ranking als Eigenvektor 4. Fragen: Exisi6ert der Eigenvektor? Usw. 5. Zweite Idee: Die Google
Unterrichtsmaterialien in digitaler und in gedruckter Form. Auszug aus:
 Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Die gentechnische Produktion von Insulin - Selbstlerneinheit zur kontextorientierten Wiederholung der molekularen Genetik Das komplette
Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Die gentechnische Produktion von Insulin - Selbstlerneinheit zur kontextorientierten Wiederholung der molekularen Genetik Das komplette
UNIVERSITÄTSKLINIKUM Schleswig-Holstein. Sequenzierung. Norbert Arnold. Dept. Gynecology and Obstetrics Oncology Laboratory
 Sequenzierung Norbert Arnold Neue Sequenziertechnologien (seit 2005) Next Generation Sequenzierung (NGS, Erste Generation) Unterschiedliche Plattformen mit unterschiedlichen Strategien Roche 454 (basierend
Sequenzierung Norbert Arnold Neue Sequenziertechnologien (seit 2005) Next Generation Sequenzierung (NGS, Erste Generation) Unterschiedliche Plattformen mit unterschiedlichen Strategien Roche 454 (basierend
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de") Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Support-Tipp Mai 2010 - Release Management in Altium Designer
 Support-Tipp Mai 2010 - Release Management in Altium Designer Mai 2010 Frage: Welche Aufgaben hat das Release Management und wie unterstützt Altium Designer diesen Prozess? Zusammenfassung: Das Glück eines
Support-Tipp Mai 2010 - Release Management in Altium Designer Mai 2010 Frage: Welche Aufgaben hat das Release Management und wie unterstützt Altium Designer diesen Prozess? Zusammenfassung: Das Glück eines
Suche schlecht beschriftete Bilder mit Eigenen Abfragen
 Suche schlecht beschriftete Bilder mit Eigenen Abfragen Ist die Bilderdatenbank über einen längeren Zeitraum in Benutzung, so steigt die Wahrscheinlichkeit für schlecht beschriftete Bilder 1. Insbesondere
Suche schlecht beschriftete Bilder mit Eigenen Abfragen Ist die Bilderdatenbank über einen längeren Zeitraum in Benutzung, so steigt die Wahrscheinlichkeit für schlecht beschriftete Bilder 1. Insbesondere
Genomsequenzierung für Anfänger
 Genomsequenzierung für Anfänger Philipp Pagel 8. November 2005 1 DNA Sequenzierung Heute wird DNA üblicherweise mit der sogenannten Sanger (oder chain-terminationoder Didesoxy-) Methode sequenziert dessen
Genomsequenzierung für Anfänger Philipp Pagel 8. November 2005 1 DNA Sequenzierung Heute wird DNA üblicherweise mit der sogenannten Sanger (oder chain-terminationoder Didesoxy-) Methode sequenziert dessen
Fachbericht zum Thema: Anforderungen an ein Datenbanksystem
 Fachbericht zum Thema: Anforderungen an ein Datenbanksystem von André Franken 1 Inhaltsverzeichnis 1 Inhaltsverzeichnis 1 2 Einführung 2 2.1 Gründe für den Einsatz von DB-Systemen 2 2.2 Definition: Datenbank
Fachbericht zum Thema: Anforderungen an ein Datenbanksystem von André Franken 1 Inhaltsverzeichnis 1 Inhaltsverzeichnis 1 2 Einführung 2 2.1 Gründe für den Einsatz von DB-Systemen 2 2.2 Definition: Datenbank
Der vorliegende Konverter unterstützt Sie bei der Konvertierung der Datensätze zu IBAN und BIC.
 Anleitung Konverter Letzte Aktualisierung dieses Dokumentes: 14.11.2013 Der vorliegende Konverter unterstützt Sie bei der Konvertierung der Datensätze zu IBAN und BIC. Wichtiger Hinweis: Der Konverter
Anleitung Konverter Letzte Aktualisierung dieses Dokumentes: 14.11.2013 Der vorliegende Konverter unterstützt Sie bei der Konvertierung der Datensätze zu IBAN und BIC. Wichtiger Hinweis: Der Konverter
SANDBOXIE konfigurieren
 SANDBOXIE konfigurieren für Webbrowser und E-Mail-Programme Dies ist eine kurze Anleitung für die grundlegenden folgender Programme: Webbrowser: Internet Explorer, Mozilla Firefox und Opera E-Mail-Programme:
SANDBOXIE konfigurieren für Webbrowser und E-Mail-Programme Dies ist eine kurze Anleitung für die grundlegenden folgender Programme: Webbrowser: Internet Explorer, Mozilla Firefox und Opera E-Mail-Programme:
Diese Ansicht erhalten Sie nach der erfolgreichen Anmeldung bei Wordpress.
 Anmeldung http://www.ihredomain.de/wp-admin Dashboard Diese Ansicht erhalten Sie nach der erfolgreichen Anmeldung bei Wordpress. Das Dashboard gibt Ihnen eine kurze Übersicht, z.b. Anzahl der Beiträge,
Anmeldung http://www.ihredomain.de/wp-admin Dashboard Diese Ansicht erhalten Sie nach der erfolgreichen Anmeldung bei Wordpress. Das Dashboard gibt Ihnen eine kurze Übersicht, z.b. Anzahl der Beiträge,
Updatehinweise für die Version forma 5.5.5
 Updatehinweise für die Version forma 5.5.5 Seit der Version forma 5.5.0 aus 2012 gibt es nur noch eine Office-Version und keine StandAlone-Version mehr. Wenn Sie noch mit der alten Version forma 5.0.x
Updatehinweise für die Version forma 5.5.5 Seit der Version forma 5.5.0 aus 2012 gibt es nur noch eine Office-Version und keine StandAlone-Version mehr. Wenn Sie noch mit der alten Version forma 5.0.x
1 topologisches Sortieren
 Wolfgang Hönig / Andreas Ecke WS 09/0 topologisches Sortieren. Überblick. Solange noch Knoten vorhanden: a) Suche Knoten v, zu dem keine Kante führt (Falls nicht vorhanden keine topologische Sortierung
Wolfgang Hönig / Andreas Ecke WS 09/0 topologisches Sortieren. Überblick. Solange noch Knoten vorhanden: a) Suche Knoten v, zu dem keine Kante führt (Falls nicht vorhanden keine topologische Sortierung
Anleitung über den Umgang mit Schildern
 Anleitung über den Umgang mit Schildern -Vorwort -Wo bekommt man Schilder? -Wo und wie speichert man die Schilder? -Wie füge ich die Schilder in meinen Track ein? -Welche Bauteile kann man noch für Schilder
Anleitung über den Umgang mit Schildern -Vorwort -Wo bekommt man Schilder? -Wo und wie speichert man die Schilder? -Wie füge ich die Schilder in meinen Track ein? -Welche Bauteile kann man noch für Schilder
Sehr geehrte Faktor-IPS Anwender,
 März 2014 Faktor-IPS 3.11 Das neue Release Faktor-IPS 3.11 steht Ihnen zum Download zur Verfügung. Wir informieren Sie über die neusten Feautres. Lesen Sie mehr Sehr geehrte Faktor-IPS Anwender, Auf faktorzehn.org
März 2014 Faktor-IPS 3.11 Das neue Release Faktor-IPS 3.11 steht Ihnen zum Download zur Verfügung. Wir informieren Sie über die neusten Feautres. Lesen Sie mehr Sehr geehrte Faktor-IPS Anwender, Auf faktorzehn.org
6. DNA -Bakteriengenetik
 6. DNA -Bakteriengenetik Konzepte: Francis Crick DNA Struktur DNA Replikation Gentransfer in Bakterien Bakteriophagen 2. Welcher der folgenden Sätze entspricht der Chargaff-Regel? A) Die Menge von Purinen
6. DNA -Bakteriengenetik Konzepte: Francis Crick DNA Struktur DNA Replikation Gentransfer in Bakterien Bakteriophagen 2. Welcher der folgenden Sätze entspricht der Chargaff-Regel? A) Die Menge von Purinen
Auswerten mit Excel. Viele Video-Tutorials auf Youtube z.b. http://www.youtube.com/watch?v=vuuky6xxjro
 Auswerten mit Excel Viele Video-Tutorials auf Youtube z.b. http://www.youtube.com/watch?v=vuuky6xxjro 1. Pivot-Tabellen erstellen: In der Datenmaske in eine beliebige Zelle klicken Registerkarte Einfügen
Auswerten mit Excel Viele Video-Tutorials auf Youtube z.b. http://www.youtube.com/watch?v=vuuky6xxjro 1. Pivot-Tabellen erstellen: In der Datenmaske in eine beliebige Zelle klicken Registerkarte Einfügen
OPERATIONEN AUF EINER DATENBANK
 Einführung 1 OPERATIONEN AUF EINER DATENBANK Ein Benutzer stellt eine Anfrage: Die Benutzer einer Datenbank können meist sowohl interaktiv als auch über Anwendungen Anfragen an eine Datenbank stellen:
Einführung 1 OPERATIONEN AUF EINER DATENBANK Ein Benutzer stellt eine Anfrage: Die Benutzer einer Datenbank können meist sowohl interaktiv als auch über Anwendungen Anfragen an eine Datenbank stellen:
Kapiteltests zum Leitprogramm Binäre Suchbäume
 Kapiteltests zum Leitprogramm Binäre Suchbäume Björn Steffen Timur Erdag überarbeitet von Christina Class Binäre Suchbäume Kapiteltests für das ETH-Leitprogramm Adressaten und Institutionen Das Leitprogramm
Kapiteltests zum Leitprogramm Binäre Suchbäume Björn Steffen Timur Erdag überarbeitet von Christina Class Binäre Suchbäume Kapiteltests für das ETH-Leitprogramm Adressaten und Institutionen Das Leitprogramm
DNA-Sequenzierung. Martina Krause
 DNA-Sequenzierung Martina Krause Inhalt Methoden der DNA-Sequenzierung Methode nach Maxam - Gilbert Methode nach Sanger Einleitung Seit 1977 stehen zwei schnell arbeitende Möglichkeiten der Sequenzanalyse
DNA-Sequenzierung Martina Krause Inhalt Methoden der DNA-Sequenzierung Methode nach Maxam - Gilbert Methode nach Sanger Einleitung Seit 1977 stehen zwei schnell arbeitende Möglichkeiten der Sequenzanalyse
Beheben von verlorenen Verknüpfungen 20.06.2005
 Vor folgender Situation ist sicher jeder Solid Edge-Anwender beim Öffnen von Baugruppen oder Drafts schon einmal gestanden: Die Ursache dafür kann sein: Die Dateien wurden über den Explorer umbenannt:
Vor folgender Situation ist sicher jeder Solid Edge-Anwender beim Öffnen von Baugruppen oder Drafts schon einmal gestanden: Die Ursache dafür kann sein: Die Dateien wurden über den Explorer umbenannt:
Informatik 12 Datenbanken SQL-Einführung
 Informatik 12 Datenbanken SQL-Einführung Gierhardt Vorbemerkungen Bisher haben wir Datenbanken nur über einzelne Tabellen kennen gelernt. Stehen mehrere Tabellen in gewissen Beziehungen zur Beschreibung
Informatik 12 Datenbanken SQL-Einführung Gierhardt Vorbemerkungen Bisher haben wir Datenbanken nur über einzelne Tabellen kennen gelernt. Stehen mehrere Tabellen in gewissen Beziehungen zur Beschreibung
Informationsgehalt von DNA
 Informationsgehalt von DNA Topics Genes code, gene organisation, signals, gene detection Genomes genome organisation, nucleotide patterns, junk DNA DNA als Informationsträger DNA Building Blocks Desoxyribose
Informationsgehalt von DNA Topics Genes code, gene organisation, signals, gene detection Genomes genome organisation, nucleotide patterns, junk DNA DNA als Informationsträger DNA Building Blocks Desoxyribose
Fotostammtisch-Schaumburg
 Der Anfang zur Benutzung der Web Seite! Alles ums Anmelden und Registrieren 1. Startseite 2. Registrieren 2.1 Registrieren als Mitglied unser Stammtischseite Wie im Bild markiert jetzt auf das Rote Register
Der Anfang zur Benutzung der Web Seite! Alles ums Anmelden und Registrieren 1. Startseite 2. Registrieren 2.1 Registrieren als Mitglied unser Stammtischseite Wie im Bild markiert jetzt auf das Rote Register
OECD Programme for International Student Assessment PISA 2000. Lösungen der Beispielaufgaben aus dem Mathematiktest. Deutschland
 OECD Programme for International Student Assessment Deutschland PISA 2000 Lösungen der Beispielaufgaben aus dem Mathematiktest Beispielaufgaben PISA-Hauptstudie 2000 Seite 3 UNIT ÄPFEL Beispielaufgaben
OECD Programme for International Student Assessment Deutschland PISA 2000 Lösungen der Beispielaufgaben aus dem Mathematiktest Beispielaufgaben PISA-Hauptstudie 2000 Seite 3 UNIT ÄPFEL Beispielaufgaben
Anmerkungen zur Übergangsprüfung
 DM11 Slide 1 Anmerkungen zur Übergangsprüfung Aufgabeneingrenzung Aufgaben des folgenden Typs werden wegen ihres Schwierigkeitsgrads oder wegen eines ungeeigneten fachlichen Schwerpunkts in der Übergangsprüfung
DM11 Slide 1 Anmerkungen zur Übergangsprüfung Aufgabeneingrenzung Aufgaben des folgenden Typs werden wegen ihres Schwierigkeitsgrads oder wegen eines ungeeigneten fachlichen Schwerpunkts in der Übergangsprüfung
Objektorientierte Programmierung für Anfänger am Beispiel PHP
 Objektorientierte Programmierung für Anfänger am Beispiel PHP Johannes Mittendorfer http://jmittendorfer.hostingsociety.com 19. August 2012 Abstract Dieses Dokument soll die Vorteile der objektorientierten
Objektorientierte Programmierung für Anfänger am Beispiel PHP Johannes Mittendorfer http://jmittendorfer.hostingsociety.com 19. August 2012 Abstract Dieses Dokument soll die Vorteile der objektorientierten
ECO-Manager - Funktionsbeschreibung
 ECO-Manager - Funktionsbeschreibung Version Autor Datum Kommentare 1.0 A. Sterzenbach 24.03.2006 - Generell Das Einarbeiten und das damit verbundene Aktualisieren eines großen Zusammenbaus (z.b. Werkzeugaufbau)
ECO-Manager - Funktionsbeschreibung Version Autor Datum Kommentare 1.0 A. Sterzenbach 24.03.2006 - Generell Das Einarbeiten und das damit verbundene Aktualisieren eines großen Zusammenbaus (z.b. Werkzeugaufbau)
Klassenentwurf. Wie schreiben wir Klassen, die leicht zu verstehen, wartbar und wiederverwendbar sind? Objektorientierte Programmierung mit Java
 Objektorientierte Programmierung mit Java Eine praxisnahe Einführung mit BlueJ Klassenentwurf Wie schreiben wir Klassen, die leicht zu verstehen, wartbar und wiederverwendbar sind? 1.0 Zentrale Konzepte
Objektorientierte Programmierung mit Java Eine praxisnahe Einführung mit BlueJ Klassenentwurf Wie schreiben wir Klassen, die leicht zu verstehen, wartbar und wiederverwendbar sind? 1.0 Zentrale Konzepte
http://train-the-trainer.fh-joanneum.at IINFO Storyboard
 IINFO Storyboard Allgemeine Bemerkungen und Richtlinien zur Handhabung. Das Storyboard besteht aus einem Web, d.h. einer vernetzten Struktur von HTML-Seiten welche später von den Programmieren direkt als
IINFO Storyboard Allgemeine Bemerkungen und Richtlinien zur Handhabung. Das Storyboard besteht aus einem Web, d.h. einer vernetzten Struktur von HTML-Seiten welche später von den Programmieren direkt als
Bioinformatische Suche nach pre-mirnas
 Bioinformatische Suche nach pre-mirnas Vorbereitung: Lesen Sie die Publikationen Meyers et al., 2008, Criteria for Annotation of Plant MicroRNAs und Thieme et al., 2011, SplamiR prediction of spliced mirnas
Bioinformatische Suche nach pre-mirnas Vorbereitung: Lesen Sie die Publikationen Meyers et al., 2008, Criteria for Annotation of Plant MicroRNAs und Thieme et al., 2011, SplamiR prediction of spliced mirnas
Dokumentation. Black- und Whitelists. Absenderadressen auf eine Blacklist oder eine Whitelist setzen. Zugriff per Webbrowser
 Dokumentation Black- und Whitelists Absenderadressen auf eine Blacklist oder eine Whitelist setzen. Zugriff per Webbrowser Inhalt INHALT 1 Kategorie Black- und Whitelists... 2 1.1 Was sind Black- und Whitelists?...
Dokumentation Black- und Whitelists Absenderadressen auf eine Blacklist oder eine Whitelist setzen. Zugriff per Webbrowser Inhalt INHALT 1 Kategorie Black- und Whitelists... 2 1.1 Was sind Black- und Whitelists?...
Albert HAYR Linux, IT and Open Source Expert and Solution Architect. Open Source professionell einsetzen
 Open Source professionell einsetzen 1 Mein Background Ich bin überzeugt von Open Source. Ich verwende fast nur Open Source privat und beruflich. Ich arbeite seit mehr als 10 Jahren mit Linux und Open Source.
Open Source professionell einsetzen 1 Mein Background Ich bin überzeugt von Open Source. Ich verwende fast nur Open Source privat und beruflich. Ich arbeite seit mehr als 10 Jahren mit Linux und Open Source.
Lineargleichungssysteme: Additions-/ Subtraktionsverfahren
 Lineargleichungssysteme: Additions-/ Subtraktionsverfahren W. Kippels 22. Februar 2014 Inhaltsverzeichnis 1 Einleitung 2 2 Lineargleichungssysteme zweiten Grades 2 3 Lineargleichungssysteme höheren als
Lineargleichungssysteme: Additions-/ Subtraktionsverfahren W. Kippels 22. Februar 2014 Inhaltsverzeichnis 1 Einleitung 2 2 Lineargleichungssysteme zweiten Grades 2 3 Lineargleichungssysteme höheren als
SJ OFFICE - Update 3.0
 SJ OFFICE - Update 3.0 Das Update auf die vorherige Version 2.0 kostet netto Euro 75,00 für die erste Lizenz. Das Update für weitere Lizenzen kostet jeweils netto Euro 18,75 (25%). inkl. Programmsupport
SJ OFFICE - Update 3.0 Das Update auf die vorherige Version 2.0 kostet netto Euro 75,00 für die erste Lizenz. Das Update für weitere Lizenzen kostet jeweils netto Euro 18,75 (25%). inkl. Programmsupport
MetaQuotes Empfehlungen zum Gebrauch von
 MetaQuotes Empfehlungen zum Gebrauch von MetaTrader 4 auf Mac OS Auch wenn viele kommerzielle Angebote im Internet existieren, so hat sich MetaQuotes, der Entwickler von MetaTrader 4, dazu entschieden
MetaQuotes Empfehlungen zum Gebrauch von MetaTrader 4 auf Mac OS Auch wenn viele kommerzielle Angebote im Internet existieren, so hat sich MetaQuotes, der Entwickler von MetaTrader 4, dazu entschieden
- Google als Suchmaschine richtig nutzen -
 - Google als Suchmaschine richtig nutzen - Google ist die wohl weltweit bekannteste und genutzte Suchmaschine der Welt. Google indexiert und aktualisiert eingetragene Seiten in bestimmten Intervallen um
- Google als Suchmaschine richtig nutzen - Google ist die wohl weltweit bekannteste und genutzte Suchmaschine der Welt. Google indexiert und aktualisiert eingetragene Seiten in bestimmten Intervallen um
ACDSee Pro 2. ACDSee Pro 2 Tutorials: Übertragung von Fotos (+ Datenbank) auf einen anderen Computer. Über Metadaten und die Datenbank
 auf einen anderen Computer. Über Metadaten und die Datenbank") Tutorials: Übertragung von Fotos (+ ) auf einen anderen Computer Export der In dieser Lektion erfahren Sie, wie Sie am effektivsten Fotos von einem Computer auf einen anderen übertragen. Wenn Sie Ihre
Tutorials: Übertragung von Fotos (+ ) auf einen anderen Computer Export der In dieser Lektion erfahren Sie, wie Sie am effektivsten Fotos von einem Computer auf einen anderen übertragen. Wenn Sie Ihre
Wie kann ich in der Backstage-Ansicht eigene Dokumentationen einbinden?
 Wie kann ich in der Backstage-Ansicht eigene Dokumentationen einbinden? Anforderung Durch die Bearbeitung einer XML-Datei können Sie Ihre eigenen Dokumentationen (z.b. PDF-Dateien, Microsoft Word Dokumente
Wie kann ich in der Backstage-Ansicht eigene Dokumentationen einbinden? Anforderung Durch die Bearbeitung einer XML-Datei können Sie Ihre eigenen Dokumentationen (z.b. PDF-Dateien, Microsoft Word Dokumente
MORE Profile. Pass- und Lizenzverwaltungssystem. Stand: 19.02.2014 MORE Projects GmbH
 MORE Profile Pass- und Lizenzverwaltungssystem erstellt von: Thorsten Schumann erreichbar unter: thorsten.schumann@more-projects.de Stand: MORE Projects GmbH Einführung Die in More Profile integrierte
MORE Profile Pass- und Lizenzverwaltungssystem erstellt von: Thorsten Schumann erreichbar unter: thorsten.schumann@more-projects.de Stand: MORE Projects GmbH Einführung Die in More Profile integrierte
2 Darstellung von Zahlen und Zeichen
 2.1 Analoge und digitale Darstellung von Werten 79 2 Darstellung von Zahlen und Zeichen Computer- bzw. Prozessorsysteme führen Transformationen durch, die Eingaben X auf Ausgaben Y abbilden, d.h. Y = f
2.1 Analoge und digitale Darstellung von Werten 79 2 Darstellung von Zahlen und Zeichen Computer- bzw. Prozessorsysteme führen Transformationen durch, die Eingaben X auf Ausgaben Y abbilden, d.h. Y = f
Datenbanken Kapitel 2
 Datenbanken Kapitel 2 1 Eine existierende Datenbank öffnen Eine Datenbank, die mit Microsoft Access erschaffen wurde, kann mit dem gleichen Programm auch wieder geladen werden: Die einfachste Methode ist,
Datenbanken Kapitel 2 1 Eine existierende Datenbank öffnen Eine Datenbank, die mit Microsoft Access erschaffen wurde, kann mit dem gleichen Programm auch wieder geladen werden: Die einfachste Methode ist,
Web-Kürzel. Krishna Tateneni Yves Arrouye Deutsche Übersetzung: Stefan Winter
 Krishna Tateneni Yves Arrouye Deutsche Übersetzung: Stefan Winter 2 Inhaltsverzeichnis 1 Web-Kürzel 4 1.1 Einführung.......................................... 4 1.2 Web-Kürzel.........................................
Krishna Tateneni Yves Arrouye Deutsche Übersetzung: Stefan Winter 2 Inhaltsverzeichnis 1 Web-Kürzel 4 1.1 Einführung.......................................... 4 1.2 Web-Kürzel.........................................
Würfelt man dabei je genau 10 - mal eine 1, 2, 3, 4, 5 und 6, so beträgt die Anzahl. der verschiedenen Reihenfolgen, in denen man dies tun kann, 60!.
 040304 Übung 9a Analysis, Abschnitt 4, Folie 8 Die Wahrscheinlichkeit, dass bei n - maliger Durchführung eines Zufallexperiments ein Ereignis A ( mit Wahrscheinlichkeit p p ( A ) ) für eine beliebige Anzahl
040304 Übung 9a Analysis, Abschnitt 4, Folie 8 Die Wahrscheinlichkeit, dass bei n - maliger Durchführung eines Zufallexperiments ein Ereignis A ( mit Wahrscheinlichkeit p p ( A ) ) für eine beliebige Anzahl
Tevalo Handbuch v 1.1 vom 10.11.2011
 Tevalo Handbuch v 1.1 vom 10.11.2011 Inhalt Registrierung... 3 Kennwort vergessen... 3 Startseite nach dem Login... 4 Umfrage erstellen... 4 Fragebogen Vorschau... 7 Umfrage fertigstellen... 7 Öffentliche
Tevalo Handbuch v 1.1 vom 10.11.2011 Inhalt Registrierung... 3 Kennwort vergessen... 3 Startseite nach dem Login... 4 Umfrage erstellen... 4 Fragebogen Vorschau... 7 Umfrage fertigstellen... 7 Öffentliche
Senden von strukturierten Berichten über das SFTP Häufig gestellte Fragen
 Senden von strukturierten Berichten über das SFTP Häufig gestellte Fragen 1 Allgemeines Was versteht man unter SFTP? Die Abkürzung SFTP steht für SSH File Transfer Protocol oder Secure File Transfer Protocol.
Senden von strukturierten Berichten über das SFTP Häufig gestellte Fragen 1 Allgemeines Was versteht man unter SFTP? Die Abkürzung SFTP steht für SSH File Transfer Protocol oder Secure File Transfer Protocol.
Bilder Schärfen und Rauschen entfernen
 Bilder Schärfen und Rauschen entfernen Um alte Bilder, so wie die von der Olympus Camedia 840 L noch dazu zu bewegen, Farben froh und frisch daherzukommen, bedarf es einiger Arbeit und die habe ich hier
Bilder Schärfen und Rauschen entfernen Um alte Bilder, so wie die von der Olympus Camedia 840 L noch dazu zu bewegen, Farben froh und frisch daherzukommen, bedarf es einiger Arbeit und die habe ich hier
Erstellung von Reports mit Anwender-Dokumentation und System-Dokumentation in der ArtemiS SUITE (ab Version 5.0)
") Erstellung von und System-Dokumentation in der ArtemiS SUITE (ab Version 5.0) In der ArtemiS SUITE steht eine neue, sehr flexible Reporting-Funktion zur Verfügung, die mit der Version 5.0 noch einmal verbessert
Erstellung von und System-Dokumentation in der ArtemiS SUITE (ab Version 5.0) In der ArtemiS SUITE steht eine neue, sehr flexible Reporting-Funktion zur Verfügung, die mit der Version 5.0 noch einmal verbessert
etutor Benutzerhandbuch XQuery Benutzerhandbuch Georg Nitsche
 etutor Benutzerhandbuch Benutzerhandbuch XQuery Georg Nitsche Version 1.0 Stand März 2006 Versionsverlauf: Version Autor Datum Änderungen 1.0 gn 06.03.2006 Fertigstellung der ersten Version Inhaltsverzeichnis:
etutor Benutzerhandbuch Benutzerhandbuch XQuery Georg Nitsche Version 1.0 Stand März 2006 Versionsverlauf: Version Autor Datum Änderungen 1.0 gn 06.03.2006 Fertigstellung der ersten Version Inhaltsverzeichnis:
Handbuch zur Anlage von Turnieren auf der NÖEV-Homepage
 Handbuch zur Anlage von Turnieren auf der NÖEV-Homepage Inhaltsverzeichnis 1. Anmeldung... 2 1.1 Startbildschirm... 3 2. Die PDF-Dateien hochladen... 4 2.1 Neue PDF-Datei erstellen... 5 3. Obelix-Datei
Handbuch zur Anlage von Turnieren auf der NÖEV-Homepage Inhaltsverzeichnis 1. Anmeldung... 2 1.1 Startbildschirm... 3 2. Die PDF-Dateien hochladen... 4 2.1 Neue PDF-Datei erstellen... 5 3. Obelix-Datei
Lokale Installation von DotNetNuke 4 ohne IIS
 Lokale Installation von DotNetNuke 4 ohne IIS ITM GmbH Wankelstr. 14 70563 Stuttgart http://www.itm-consulting.de Benjamin Hermann hermann@itm-consulting.de 12.12.2006 Agenda Benötigte Komponenten Installation
Lokale Installation von DotNetNuke 4 ohne IIS ITM GmbH Wankelstr. 14 70563 Stuttgart http://www.itm-consulting.de Benjamin Hermann hermann@itm-consulting.de 12.12.2006 Agenda Benötigte Komponenten Installation
Theoretische Grundlagen der Informatik WS 09/10
 Theoretische Grundlagen der Informatik WS 09/10 - Tutorium 6 - Michael Kirsten und Kai Wallisch Sitzung 13 02.02.2010 Inhaltsverzeichnis 1 Formeln zur Berechnung Aufgabe 1 2 Hamming-Distanz Aufgabe 2 3
Theoretische Grundlagen der Informatik WS 09/10 - Tutorium 6 - Michael Kirsten und Kai Wallisch Sitzung 13 02.02.2010 Inhaltsverzeichnis 1 Formeln zur Berechnung Aufgabe 1 2 Hamming-Distanz Aufgabe 2 3
So die eigene WEB-Seite von Pinterest verifizieren lassen!
 So die eigene WEB-Seite von Pinterest verifizieren lassen! Quelle: www.rohinie.eu Die eigene Seite auf Pinterest verifizieren Es ist offiziell. Vielleicht haben auch Sie in den vergangenen Wochen die Informationen
So die eigene WEB-Seite von Pinterest verifizieren lassen! Quelle: www.rohinie.eu Die eigene Seite auf Pinterest verifizieren Es ist offiziell. Vielleicht haben auch Sie in den vergangenen Wochen die Informationen
Installation der SAS Foundation Software auf Windows
 Installation der SAS Foundation Software auf Windows Der installierende Benutzer unter Windows muss Mitglied der lokalen Gruppe Administratoren / Administrators sein und damit das Recht besitzen, Software
Installation der SAS Foundation Software auf Windows Der installierende Benutzer unter Windows muss Mitglied der lokalen Gruppe Administratoren / Administrators sein und damit das Recht besitzen, Software
Übung II. Einführung, Teil 1. Arbeiten mit Ensembl
 Übung II Einführung, Teil 1 Arbeiten mit Ensembl Ensembl Genome Browser (Bereitstellung von Vielzeller Genomen) Projekt wurde 1999 initiiert Projektpartner EMBL European Bioinformatics Institute (EBI)
Übung II Einführung, Teil 1 Arbeiten mit Ensembl Ensembl Genome Browser (Bereitstellung von Vielzeller Genomen) Projekt wurde 1999 initiiert Projektpartner EMBL European Bioinformatics Institute (EBI)
Access 2013. Grundlagen für Anwender. Susanne Weber. 1. Ausgabe, 1. Aktualisierung, Juni 2013
 Access 2013 Susanne Weber 1. Ausgabe, 1. Aktualisierung, Juni 2013 Grundlagen für Anwender ACC2013 2 Access 2013 - Grundlagen für Anwender 2 Mit Datenbanken arbeiten In diesem Kapitel erfahren Sie was
Access 2013 Susanne Weber 1. Ausgabe, 1. Aktualisierung, Juni 2013 Grundlagen für Anwender ACC2013 2 Access 2013 - Grundlagen für Anwender 2 Mit Datenbanken arbeiten In diesem Kapitel erfahren Sie was
Theoretische Grundlagen der Informatik
 Theoretische Grundlagen der Informatik Vorlesung am 12.01.2012 INSTITUT FÜR THEORETISCHE 0 KIT 12.01.2012 Universität des Dorothea Landes Baden-Württemberg Wagner - Theoretische und Grundlagen der Informatik
Theoretische Grundlagen der Informatik Vorlesung am 12.01.2012 INSTITUT FÜR THEORETISCHE 0 KIT 12.01.2012 Universität des Dorothea Landes Baden-Württemberg Wagner - Theoretische und Grundlagen der Informatik
Abamsoft Finos im Zusammenspiel mit shop to date von DATA BECKER
 Abamsoft Finos im Zusammenspiel mit shop to date von DATA BECKER Abamsoft Finos in Verbindung mit der Webshopanbindung wurde speziell auf die Shop-Software shop to date von DATA BECKER abgestimmt. Mit
Abamsoft Finos im Zusammenspiel mit shop to date von DATA BECKER Abamsoft Finos in Verbindung mit der Webshopanbindung wurde speziell auf die Shop-Software shop to date von DATA BECKER abgestimmt. Mit
4D Server v12 64-bit Version BETA VERSION
 4D Server v12 64-bit Version BETA VERSION 4D Server v12 unterstützt jetzt das Windows 64-bit Betriebssystem. Hauptvorteil der 64-bit Technologie ist die rundum verbesserte Performance der Anwendungen und
4D Server v12 64-bit Version BETA VERSION 4D Server v12 unterstützt jetzt das Windows 64-bit Betriebssystem. Hauptvorteil der 64-bit Technologie ist die rundum verbesserte Performance der Anwendungen und
Mit der Maus im Menü links auf den Menüpunkt 'Seiten' gehen und auf 'Erstellen klicken.
 Seite erstellen Mit der Maus im Menü links auf den Menüpunkt 'Seiten' gehen und auf 'Erstellen klicken. Es öffnet sich die Eingabe Seite um eine neue Seite zu erstellen. Seiten Titel festlegen Den neuen
Seite erstellen Mit der Maus im Menü links auf den Menüpunkt 'Seiten' gehen und auf 'Erstellen klicken. Es öffnet sich die Eingabe Seite um eine neue Seite zu erstellen. Seiten Titel festlegen Den neuen
Wie gestaltet man Online-Umfragen mit SurveyMonkey?
 Wie gestaltet man Online-Umfragen mit SurveyMonkey? 1. Auf www.surveymonkey.com gehen. Zu allererst muss man sich registrieren. Auf der linken Seite auf Join now for free klicken. 2. Maske ausfüllen und
Wie gestaltet man Online-Umfragen mit SurveyMonkey? 1. Auf www.surveymonkey.com gehen. Zu allererst muss man sich registrieren. Auf der linken Seite auf Join now for free klicken. 2. Maske ausfüllen und
Erstellen eigener HTML Seiten auf ewon
 ewon - Technical Note Nr. 010 Version 1.2 Erstellen eigener HTML Seiten auf ewon 30.08.2006/SI Übersicht: 1. Thema 2. Benötigte Komponenten 3. Funktionsaufbau und Konfiguration 3.1. Unterpunkt 1 3.2. Unterpunkt
ewon - Technical Note Nr. 010 Version 1.2 Erstellen eigener HTML Seiten auf ewon 30.08.2006/SI Übersicht: 1. Thema 2. Benötigte Komponenten 3. Funktionsaufbau und Konfiguration 3.1. Unterpunkt 1 3.2. Unterpunkt
PTV VISWALK TIPPS UND TRICKS PTV VISWALK TIPPS UND TRICKS: VERWENDUNG DICHTEBASIERTER TEILROUTEN
 PTV VISWALK TIPPS UND TRICKS PTV VISWALK TIPPS UND TRICKS: VERWENDUNG DICHTEBASIERTER TEILROUTEN Karlsruhe, April 2015 Verwendung dichte-basierter Teilrouten Stellen Sie sich vor, in einem belebten Gebäude,
PTV VISWALK TIPPS UND TRICKS PTV VISWALK TIPPS UND TRICKS: VERWENDUNG DICHTEBASIERTER TEILROUTEN Karlsruhe, April 2015 Verwendung dichte-basierter Teilrouten Stellen Sie sich vor, in einem belebten Gebäude,
Dokumentation für das Checkoutsystem von Freshworx
 Dokumentation für das Checkoutsystem von Freshworx Auf den folgenden Seiten finden Sie eine ausführliche Anleitung zur Einrichtung des Checkoutsystems von Freshworx. Sollten Sie Probleme bei der Einrichtung
Dokumentation für das Checkoutsystem von Freshworx Auf den folgenden Seiten finden Sie eine ausführliche Anleitung zur Einrichtung des Checkoutsystems von Freshworx. Sollten Sie Probleme bei der Einrichtung
Problem crazytrickler unter Windows 8:
 Problem crazytrickler unter Windows 8: Für die Kommunikation mit dem PC ist im crazytrickler der Chip PL2303HXA/XA zuständig. Er wird unter Windows 8 nicht mehr vom Hersteller Prolific unterstützt. Geräte
Problem crazytrickler unter Windows 8: Für die Kommunikation mit dem PC ist im crazytrickler der Chip PL2303HXA/XA zuständig. Er wird unter Windows 8 nicht mehr vom Hersteller Prolific unterstützt. Geräte
trivum Multiroom System Konfigurations- Anleitung Erstellen eines RS232 Protokolls am Bespiel eines Marantz SR7005
 trivum Multiroom System Konfigurations- Anleitung Erstellen eines RS232 Protokolls am Bespiel eines Marantz SR7005 2 Inhalt 1. Anleitung zum Einbinden eines über RS232 zu steuernden Devices...3 1.2 Konfiguration
trivum Multiroom System Konfigurations- Anleitung Erstellen eines RS232 Protokolls am Bespiel eines Marantz SR7005 2 Inhalt 1. Anleitung zum Einbinden eines über RS232 zu steuernden Devices...3 1.2 Konfiguration
Die Excel Schnittstelle - Pro Pack
 Die Excel Schnittstelle - Pro Pack Die Excel Pro Pack ist eine Erweiterung der normalen Excel Schnittstelle, die in der Vollversion von POSWare Bestandteil der normalen Lizenz und somit für alle Lizenznehmer
Die Excel Schnittstelle - Pro Pack Die Excel Pro Pack ist eine Erweiterung der normalen Excel Schnittstelle, die in der Vollversion von POSWare Bestandteil der normalen Lizenz und somit für alle Lizenznehmer
Binäre Bäume. 1. Allgemeines. 2. Funktionsweise. 2.1 Eintragen
 Binäre Bäume 1. Allgemeines Binäre Bäume werden grundsätzlich verwendet, um Zahlen der Größe nach, oder Wörter dem Alphabet nach zu sortieren. Dem einfacheren Verständnis zu Liebe werde ich mich hier besonders
Binäre Bäume 1. Allgemeines Binäre Bäume werden grundsätzlich verwendet, um Zahlen der Größe nach, oder Wörter dem Alphabet nach zu sortieren. Dem einfacheren Verständnis zu Liebe werde ich mich hier besonders
Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS Yvonne Lichtblau/Johannes Starlinger
 Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS 2017 Yvonne Lichtblau/Johannes Starlinger Presentations Assignment 2 Yvonne Lichtblau Übungen Grundlagen der Bioinformatik SS 2017
Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS 2017 Yvonne Lichtblau/Johannes Starlinger Presentations Assignment 2 Yvonne Lichtblau Übungen Grundlagen der Bioinformatik SS 2017
Unterrichtsmaterialien in digitaler und in gedruckter Form. Auszug aus: Übungsbuch für den Grundkurs mit Tipps und Lösungen: Analysis
 Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Übungsbuch für den Grundkurs mit Tipps und Lösungen: Analysis Das komplette Material finden Sie hier: Download bei School-Scout.de
Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Übungsbuch für den Grundkurs mit Tipps und Lösungen: Analysis Das komplette Material finden Sie hier: Download bei School-Scout.de
3 Windows als Storage-Zentrale
 3 Windows als Storage-Zentrale Windows als zentrale Datenspeichereinheit punktet gegenüber anderen Lösungen vor allem bei der Integration in vorhandene Unternehmensnetze sowie bei der Administration. Dabei
3 Windows als Storage-Zentrale Windows als zentrale Datenspeichereinheit punktet gegenüber anderen Lösungen vor allem bei der Integration in vorhandene Unternehmensnetze sowie bei der Administration. Dabei
SEPA-Anleitung zum Release 3.09
 Hier folgt nun eine kurze Information was sich mit dem neuen Release 3.08 zum Thema SEPA alles ändert. Bitte diese Anleitung sorgfältig lesen, damit bei der Umsetzung keine Fragen aufkommen. Bitte vor
Hier folgt nun eine kurze Information was sich mit dem neuen Release 3.08 zum Thema SEPA alles ändert. Bitte diese Anleitung sorgfältig lesen, damit bei der Umsetzung keine Fragen aufkommen. Bitte vor
Bauteilattribute als Sachdaten anzeigen
 Mit den speedikon Attributfiltern können Sie die speedikon Attribute eines Bauteils als MicroStation Sachdaten an die Elemente anhängen Inhalte Was ist ein speedikon Attribut?... 3 Eigene Attribute vergeben...
Mit den speedikon Attributfiltern können Sie die speedikon Attribute eines Bauteils als MicroStation Sachdaten an die Elemente anhängen Inhalte Was ist ein speedikon Attribut?... 3 Eigene Attribute vergeben...
Übungsblatt: Protein interaction networks. Ulf Leser and Samira Jaeger
 Übungsblatt: Protein interaction networks Ulf Leser and Samira Jaeger Aufgabe 1 Netzwerkzentralität (6P) In der Vorlesung haben Degree Centrality besprochen. Finde drei weitere etablierte Zentralitätsmaße
Übungsblatt: Protein interaction networks Ulf Leser and Samira Jaeger Aufgabe 1 Netzwerkzentralität (6P) In der Vorlesung haben Degree Centrality besprochen. Finde drei weitere etablierte Zentralitätsmaße
Aminosäuren - Proteine
 Aminosäuren - Proteine ÜBERBLICK D.Pflumm KSR / MSE Aminosäuren Überblick Allgemeine Formel für das Grundgerüst einer Aminosäure Carboxylgruppe: R-COOH O Aminogruppe: R-NH 2 einzelnes C-Atom (α-c-atom)
Aminosäuren - Proteine ÜBERBLICK D.Pflumm KSR / MSE Aminosäuren Überblick Allgemeine Formel für das Grundgerüst einer Aminosäure Carboxylgruppe: R-COOH O Aminogruppe: R-NH 2 einzelnes C-Atom (α-c-atom)
S/W mit PhotoLine. Inhaltsverzeichnis. PhotoLine
 PhotoLine S/W mit PhotoLine Erstellt mit Version 16.11 Ich liebe Schwarzweiß-Bilder und schaue mir neidisch die Meisterwerke an, die andere Fotografen zustande bringen. Schon lange versuche ich, auch so
PhotoLine S/W mit PhotoLine Erstellt mit Version 16.11 Ich liebe Schwarzweiß-Bilder und schaue mir neidisch die Meisterwerke an, die andere Fotografen zustande bringen. Schon lange versuche ich, auch so
mobilepoi 0.91 Demo Version Anleitung Das Software Studio Christian Efinger Erstellt am 21. Oktober 2005
 Das Software Studio Christian Efinger mobilepoi 0.91 Demo Version Anleitung Erstellt am 21. Oktober 2005 Kontakt: Das Software Studio Christian Efinger ce@efinger-online.de Inhalt 1. Einführung... 3 2.
Das Software Studio Christian Efinger mobilepoi 0.91 Demo Version Anleitung Erstellt am 21. Oktober 2005 Kontakt: Das Software Studio Christian Efinger ce@efinger-online.de Inhalt 1. Einführung... 3 2.
tentoinfinity Apps 1.0 EINFÜHRUNG
 tentoinfinity Apps Una Hilfe Inhalt Copyright 2013-2015 von tentoinfinity Apps. Alle Rechte vorbehalten. Inhalt der online-hilfe wurde zuletzt aktualisiert am August 6, 2015. Zusätzlicher Support Ressourcen
tentoinfinity Apps Una Hilfe Inhalt Copyright 2013-2015 von tentoinfinity Apps. Alle Rechte vorbehalten. Inhalt der online-hilfe wurde zuletzt aktualisiert am August 6, 2015. Zusätzlicher Support Ressourcen
Wie Sie mit Mastern arbeiten
 Wie Sie mit Mastern arbeiten Was ist ein Master? Einer der großen Vorteile von EDV besteht darin, dass Ihnen der Rechner Arbeit abnimmt. Diesen Vorteil sollten sie nutzen, wo immer es geht. In PowerPoint
Wie Sie mit Mastern arbeiten Was ist ein Master? Einer der großen Vorteile von EDV besteht darin, dass Ihnen der Rechner Arbeit abnimmt. Diesen Vorteil sollten sie nutzen, wo immer es geht. In PowerPoint