Von der anwenderbezogenen Beschaffenheit der Verpackung zur Regulatory Excellence: Designplanung und Verifikation
|
|
|
- Sofie Irmela Albert
- vor 7 Jahren
- Abrufe
Transkript
1 Von der anwenderbezogenen Beschaffenheit der Verpackung zur Regulatory Excellence: Designplanung und Verifikation Franz Döpp, Senior Regulatory Affairs Manager Veranstalter: Institut für Medizintechnik Knowledge Reutlingen University
2 Wir schaffen Marktzugang für Medizinprodukte Metecon ist strategischer Partner für Medizintechnikunternehmen über den GESAMTEN Produktlebenszyklus. Wir hören zu, denken mit und schaffen individuelle Lösungen für die konforme internationale Vermarktung von Medizinprodukten.
3 Agenda 1 2 Motivation und Auftakt Übersicht der aktuellen und zukünftigen Anforderungen Sinn und Zweck der Verpackung 3 Design, Validierungsplan und Verifikationsstrategie 4 Kennzeichnung der Verpackung 5 Zusammenfassung, Lernkontrolle, Fragen
4 Unveränderte Ist-Situation in Deutschland 6 MPG Medizinprodukte ( ) dürfen in Deutschland nur in den Verkehr gebracht oder in Betrieb genommen werden, wenn sie mit der CE-Kennzeichnung nach Maßgabe der Absätze 2 Satz 1 und 3 Satz 1 versehen sind.
5 Grundregel Voraussetzung der CE-Kennzeichnung Erfüllung der Grundlegenden Anforderungen Durchführung des Konformitätsbewertungsverfahren
6 Konformitätsvermutung durch harm.norm CE-Kennzeichen: Nachweis Sicherheit + Funktion (entsprechend der Zweckbestimmung) + Erfüllung der gesetzlichen Anforderungen (Anhang I, 93/42/EWG) Design- und Entwicklungsverifizierung Präklinik: Dimensionen Reinheit Biokompatibilität Sterilität Unversehrtheit Elektrische Sicherheit QM-System (ISO 13485) Design- und Entwicklungsvalidierung Klinik: Klinische Prüfung Klinische Bewertung Ggf. Handhabung/ Gebrauchstauglichkeit

7 Grundwirkungsprinzip und solide Basis Aufzeichnungen Harmonisierte Normen EN ISO EN ISO 1041 EN ISO / 980 / 780 EN ISO QMS / IMS Dokumentieren, Lenken, Archivieren Tracability Reduzierung der Komplexität Umsetzung der Good-Practices -GPSG, ProdHaftG, BGB, HGB -MPG und Verordnungen 93/42/EWG ; 2007/47/EG 2002/95/EG; 2005/96/EG; 2011/65/EU 2006/95/EG; 2004/108/EC; 1999/5/EG Analyse der nationalen und zielmarktspezifischen Anforderungen Nachhaltige und nachvollziehbare Planung der Produktrealisierung Aufbau Audit- und Nachweispfad von relevanten Regularien zu eigener Welt
8 EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Sicherheits- und Leistungsanforderungen an ein Medizinprodukt Komplett neuer Aufbau und Neuordnung der Kapitel und Anhänge Geänderte Klassifizierungsregeln Es wird ggf. zu Höherklassifizierungen kommen mit z.t. drastischen Ausmaßen
9 EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Klinische Bewertung Konformitätsbewertungsverfahren Ggf. Scrunity UDI Es wird eine EU-Verordnung, welche sofort in Kraft tritt. Übergangszeit 3 Jahre
10 Sinn und Zweck der Verpackung
11 Wertvolle Aufgaben der Verpackung
12 Sinn und Zweck der Verpackung Funktion Schutz Lagerung Transport Kommissionierung, Manipulation Information Werbung, Marketing Zusatz Aussehen Erhöhung der Verkaufsfähigkeit Vereinbarte Beschaffenheit

13 Gesetzliche und vertragliche Rückgriffsansprüche Kaufvertrag 433 Verkäufer Garantie Kaufvertrag Hersteller Produkt- und Kunde Produzentenhaftung Rückgriff Lieferkette Zulieferer
14 EN ISO Beschaffungsprozess Die Organisation muss dokumentierte Verfahren einführen, um sicherzustellen, dass die beschafften Produkte die festgelegten Beschaffungsanforderungen erfüllen Wareneingangsprüfungen, Qualitätsendprüfungen beim Lieferanten, Erstbemusterung
15 EN ISO Validierung der Prozesse zur Produktion und zur Dienstleistungserbringung Die Organisation muss sämtliche Prozesse der Produktion und Dienstleistungserbringung validieren... Dies betrifft auch alle Prozesse, bei denen sich Unzulänglichkeiten erst zeigen, nachdem das Produkt in Gebrauch gekommen oder die Dienstleistung erbracht worden ist.
16 EN ISO Kapitel 6 Management von Resourcen Aktualisierte Forderungen zu... Vorbeugende Wartung Personal- und Umgebungshygiene Lenkung besonderer Umgebungsbedingungen... Ihre Aufgaben Festlegung der Verantwortung und Fähigkeiten des Personals, welche die Produktqualität beeinflusst Ressourcen ermitteln und bereitstellen Dokumentation von Instandhaltungsarbeiten, Lenkung der Aufzeichnungen
17 Design, Validierungsplan und Verifikationsstrategie Nachweis der gewünschten Wirkung
18 Ressourcenschonende Lösungsansätze Notwendiges klären: Spezifikation des notwendigen Produktschutzes Usability Aspekte, Identifikation der aktuellen und zukünftigen Vertriebsländer für die Produkte Herausarbeiten der regulatorischen Anforderungen bzgl. Normen und nationalen Vorgaben, Festlegung Akzeptanzkriterien, Assurance Level Ausloten der Supply-Chain und Konkretisierung der Distributionskette Ermittlung durchschnittlicher und zugesicherten Transportdauern Ableitung Worst-Case Scenarios aus Produkt-Design Abschluss Design-Phase Verpackung
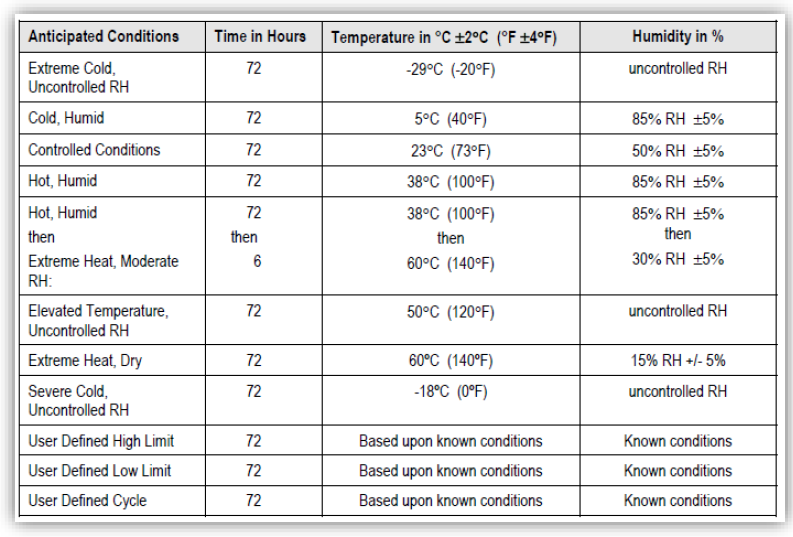

19 Verifizierungsplan
20 Kennzeichnung der Verpackung Transport- und Produktverpackung

21 Organisationspflichten Organisationspflichten Konstruktion Fabrikation Instruktion Marktbeobachtung
22 Instruktionspflichten Ausreichende Belehrung der Produktbenutzer durch den Hersteller (z.b. Warnhinweise) über mögliche Gefahrenquellen über die Grenzen der Produktanwendung über Gefahren bei naheliegender missbräuchlicher Produktverwendung, die sich noch im Rahmen der allgemeinen Zweckbestimmung des Produktes hält
23 MDD Anhang I: Grundlegende Anforderungen Kapitel 13 Bereitstellung von Informationen durch den Hersteller 13.1 Allgemeine Anforderungen 13.2 Angaben in Form von Symbolen 13.3 Kennzeichnung 13.4 Anforderung an Produkte ohne offensichtliche Zweckbestimmung 13.5 Identifizierbarkeit von Produkten und abnehmbaren Bauteilen 13.6 Gebrauchsanweisung
24 MDD Anhang I: 13.2 Angaben in Form von Symbolen MediFirma Max-Muster-Str Musterstadt
 Rückkopplung der")
25 Standard-Versandetikett (Auszug, Beispiel) Rückkopplung der Verifizierungsergebnisse


26 Haben Sie Fragen? Franz Döpp Senior Regulatory Affairs Manager Metecon GmbH Tel.: / Fax: / Mobil: /
27 Anhang zum Nachlesen Wenn Sie weitere Hintergründe erfahren möchten
28 Mögliche Rechtsfolgen aus öffentlich-rechtlicher Haftung (Auszug) 1 I ProdHaftG: Wird durch den Fehler eines Produktes ein Mensch getötet, sein Körper oder seine Gesundheit verletzt oder eine Sache beschädigt, so ist der Hersteller des Produktes verpflichtet, dem Geschädigten den daraus entstandenen Schaden zu ersetzen. 4 I ProdHaftG: Hersteller ist, wer das Endprodukt, einen Grundstoff oder ein Teilprodukt hergestellt hat. Als Hersteller gilt auch jeder, der sich durch Anbringen seines Namens, seiner Marke oder eines anderen unterscheidungskräftigen Kennzeichens als Hersteller ausgibt. 4 II ProdHaftG: Als Hersteller gilt ferner, wer ein Produkt zum Zwecke des Verkaufs ( ) im Rahmen seiner geschäftlichen Tätigkeit in den Geltungsbereich des Abkommens über den Europäischen Wirtschaftsraum einführt oder verbringt.
29 Abwehr von Schadensersatzansprüchen Zweckbestimmung eingehalten? Weicht der Betreiber oder der Anwender eines Medizinprodukts von der Zweckbestimmung ab, stellt dies ein eigenverantwortliches Handeln in Verbindung mit der Übernahme der vollständigen Haftung nicht nur für die geänderte Anwendung dar. Folglich haftet der Hersteller bei Produktmängeln ausschließlich im Rahmen der von ihm festgelegten Zweckbestimmung sowie dem vorhersehbaren Missbrauch Voraussetzung: Anwendung und objektiver Beweis für die Erfüllung der DIN EN ISO 14971; DIN EN ISO 62366; DIN EN ISO Klar erkennbare Gebrauchsanweisung gemäß dem Stand der Technik
30 MDD Anhang I: 13.3 Angaben der Kennzeichnung Name oder Firma und Anschrift des Herstellers und ggf. des EU-Repräsentanten Angaben, aus denen ersichtlich ist, um was es sich bei dem Produkt / Packungsinhalt handelt
31 MDD Anhang I: 13.3 Angaben der Kennzeichnung ggf. der Hinweis STERIL mit Angabe des Sterilisationsverfahrens Falls ein Produkt vom gleichen Hersteller auch nicht steril erhältlich ist und Verwechslungsgefahr besteht, muss evtl. auch auf Nichtsterilität hingewiesen werden ggf. das angewendete Sterilisationsverfahren ggf. Hinweis auf ein Derivat aus menschlichem Blut
32 Häufigste Fehler in der Praxis Kein Validierungsplan Keine Übersicht der geplanten und durchgeführten Verifizierungen Keine Rückkopplung zu - Risk Management File bzw. Aktivitäten - Reklamationen - CAPAS - Change Control / Chance Request & Approval Hersteller hat kein etabliertes Verfahren für die Transportvalidierung und Validierungsmasterplan
33 Regulatorische Anforderungen / Leitlinien und Normen Gesetze und Verordnungen MPG: Medizinproduktegesetz Distribution simulation testing ASTM D4169 ISTA 2A Product Performance ISO Accelerated Aging ASTM F1980 Produktspezifische Normen Horizontale Vorgaben: ZLG z.b. ISO : Intraokularlinsen, Haltbarkeits- und Transportprüfungen ISO : Ophthalmische Instrumente Grundlegende Anforderungen und Prüfverfahren RDS : Mindestinhalte von Validierungsberichten gemäß DIN EN ISO für die Sterilisation mit Ethylenoxid
34 Auswirkungen der Änderungen für den EWR Kompletter Neuaufbau der Produktakte notwendig Neufassung der Checkliste für Allgemeine Sicherheits- und Leistungsanforderungen (ehemals Grundlegende Anforderungen) Noch größerer Detaillierungsgrad UDI einführen Vorher: Stammdaten bereinigen, Produkthirarchie klären/definieren. Stakeholder einbeziehen Verifizierungs- und Validierungsergebnisse aktualisieren u.a. für Druckund Scanprozesse Erstellung/Aktualisierung der IFUs u.a.wg. krebserregenden und erbgutverändernden Stoffen & UDI; Vorkommnisse Klinische Bewertung wahrscheinlich dringlich zu aktualisieren inkl. SOPs. Erhöhte Anforderungen auch an Benannte Stellen
35 Klinische Bewertung - Klinische Daten Bereits seit 2010 gültig: Klinische Daten: Sicherheits- und/ oder Leistungsangaben, welche aus der Verwendung eines Produktes hervorgehen Der Nachweis der Übereinstimmung mit den grundlegenden Anforderungen muss eine klinische Bewertung gemäß Anhang X umfassen (vor dem ersten Inverkehrbringen)! Kritische Bewertung der wissenschaftlichen Fachliteratur Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts: über Nachweis der Gleichartigkeit des Produkts mit dem Produkt, auf das sich die Daten beziehen Kritische Bewertung der Ergebnisse sämtlicher durchgeführten klinischen Prüfungen des betreffenden Produkts ODER: Kritische Bewertung der kombinierten klinischen Daten aus wissenschaftlicher Fachliteratur und klinischen Prüfungen
Klinische Bewertung & Marktbeobachtung
 Klinische Bewertung & Marktbeobachtung, Senior Regulatory Affairs Manager EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Sicherheits- und Leistungsanforderungen an ein Medizinprodukt
Klinische Bewertung & Marktbeobachtung, Senior Regulatory Affairs Manager EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Sicherheits- und Leistungsanforderungen an ein Medizinprodukt
Know-How für die Medizintechnik
 Know-How für die Medizintechnik ISO 13485:2016 - Neue Anforderungen Qualitätsmanagement für Medizinprodukte LISAvienna Business Treff Medizintechnik Regulatory Update 2016 am 07.06.2016 in Wien DI Martin
Know-How für die Medizintechnik ISO 13485:2016 - Neue Anforderungen Qualitätsmanagement für Medizinprodukte LISAvienna Business Treff Medizintechnik Regulatory Update 2016 am 07.06.2016 in Wien DI Martin
ISO 13485:2016. TÜV SÜD Akademie GmbH Rev. <xx > / Stand: EN ISO 13485:2016 1
 ISO 13485:2016 Änderungen / Neue Anforderungen TÜV SÜD Akademie GmbH Rev. / Stand: 07 2016 EN ISO 13485:2016 1 ISO 13485:2016 Änderungen / Neue Anforderungen EN ISO 13485 DIN EN ISO 13485 TÜV SÜD
ISO 13485:2016 Änderungen / Neue Anforderungen TÜV SÜD Akademie GmbH Rev. / Stand: 07 2016 EN ISO 13485:2016 1 ISO 13485:2016 Änderungen / Neue Anforderungen EN ISO 13485 DIN EN ISO 13485 TÜV SÜD
Revision RL 93/42/EWG Aufgaben der Benannten Stelle
 RICHTLINIE 2007/47/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. September 2007 zur Änderung der Richtlinien 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über aktive
RICHTLINIE 2007/47/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. September 2007 zur Änderung der Richtlinien 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über aktive
Know-How für Medizinprodukte
 Know-How für Medizinprodukte ISO 13485:2016: Auswirkungen & Empfehlungen für die Umstellung LISAvienna Business Treff: Die neuen Herausforderungen für Medizinprodukte & IVD am 07.11.2017 in Wien DI Martin
Know-How für Medizinprodukte ISO 13485:2016: Auswirkungen & Empfehlungen für die Umstellung LISAvienna Business Treff: Die neuen Herausforderungen für Medizinprodukte & IVD am 07.11.2017 in Wien DI Martin
CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess
 CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess IHK Dialog Gesundheitswirtschaft: Medizintechnik in der Region Köln-Bonn 14. Februar 2017 2017 by qcmed GmbH, Dr. Carola
CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess IHK Dialog Gesundheitswirtschaft: Medizintechnik in der Region Köln-Bonn 14. Februar 2017 2017 by qcmed GmbH, Dr. Carola
MMedizintechnik. Artikel 12 MDD. nagement. Kombination von Medizinprodukten (Leitfaden Artikel 12 MDD) POSITION. Richtlinie 93/42/EWG.
 POSITION. Richtlinie 93/42/EWG.") Leitfaden Kombination von Medizinprodukten (Leitfaden Artikel 12 MDD) Regulatorische Anforderungen Richtlinie 93/42/EWG MMedizintechnik Artikel 12 MDD Haftungsfragen nagement POSITION Februar 2018 Zentralverband
Leitfaden Kombination von Medizinprodukten (Leitfaden Artikel 12 MDD) Regulatorische Anforderungen Richtlinie 93/42/EWG MMedizintechnik Artikel 12 MDD Haftungsfragen nagement POSITION Februar 2018 Zentralverband
Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen
 Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen build.well.being 2017 Andreas Böhler R n B Medical Software Consulting GmbH office@rnb-consulting.at build.well.being 2017 30.
Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen build.well.being 2017 Andreas Böhler R n B Medical Software Consulting GmbH office@rnb-consulting.at build.well.being 2017 30.
Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz. SPECTARIS-Infoveranstaltung
 Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz SPECTARIS-Infoveranstaltung München, 10. Januar 2014 Was ist zu tun? - Warum ist meine Brille ein Medizinprodukt? - Welche Maßnahmen muss ich
Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz SPECTARIS-Infoveranstaltung München, 10. Januar 2014 Was ist zu tun? - Warum ist meine Brille ein Medizinprodukt? - Welche Maßnahmen muss ich
Übersicht über ISO 9001:2000
 Übersicht über die ISO 9001:2000 0 Einleitung 1 Anwendungsbereich 2 Normative Verweisungen 3 Begriffe Übersicht über die ISO 9001:2000 4 Qualitätsmanagementsystem 5 Verantwortung der Leitung 6 Management
Übersicht über die ISO 9001:2000 0 Einleitung 1 Anwendungsbereich 2 Normative Verweisungen 3 Begriffe Übersicht über die ISO 9001:2000 4 Qualitätsmanagementsystem 5 Verantwortung der Leitung 6 Management
Die Kombination von Medizinprodukten. SystemCheck
 Die Kombination von Medizinprodukten SystemCheck Fachtagung der FKT 12.06.2008 Untertitel Die Prüfung und Bewertung von medizinischen elektrischen Systemen mit rechtssicherer Dokumentation zum Schutz von
Die Kombination von Medizinprodukten SystemCheck Fachtagung der FKT 12.06.2008 Untertitel Die Prüfung und Bewertung von medizinischen elektrischen Systemen mit rechtssicherer Dokumentation zum Schutz von
Klinische Prüfung von Medizinprodukten in Deutschland
 Klinische Prüfung von Medizinprodukten in Deutschland Notwendigkeit und Anforderungen 9. DVMD-Fachtagung, 31.03.2006, Erlangen Anne Eichberger 3M 2006 1 Anne Eichberger 30.03.2006 Was ist ein Medizinprodukt?
Klinische Prüfung von Medizinprodukten in Deutschland Notwendigkeit und Anforderungen 9. DVMD-Fachtagung, 31.03.2006, Erlangen Anne Eichberger 3M 2006 1 Anne Eichberger 30.03.2006 Was ist ein Medizinprodukt?
Konformität in der Apheresetechnik. Uwe Wallstab Roland E. Winkler Wolfgang Ramlow
 Konformität in der Apheresetechnik Uwe Wallstab Roland E. Winkler Wolfgang Ramlow Institut für Angewandte Biowissenschaften e.v., 1999 Konformitätsbewertung in der Apherese- Technik Wichtige Rechtsvorschriften
Konformität in der Apheresetechnik Uwe Wallstab Roland E. Winkler Wolfgang Ramlow Institut für Angewandte Biowissenschaften e.v., 1999 Konformitätsbewertung in der Apherese- Technik Wichtige Rechtsvorschriften
Präsentator. Markus Frei. avasis AG 9442 Berneck 2. Geschäftsführer
 ISO13485:2016 1 Präsentator Markus Frei Geschäftsführer avasis AG 9442 Berneck 2 Wer / Was ist medplm? avasis Siemens Industry Software 3 Dieses Webinar dauert ca. 30 Minuten zeigt die Teilnehmer nicht
ISO13485:2016 1 Präsentator Markus Frei Geschäftsführer avasis AG 9442 Berneck 2 Wer / Was ist medplm? avasis Siemens Industry Software 3 Dieses Webinar dauert ca. 30 Minuten zeigt die Teilnehmer nicht
Interpretation der Anforderungen
 Interpretation der Anforderungen Canisha Teubert Online-Handbücher für Technik, IT, Produktentwicklung Medizinprodukte planen, entwickeln, realisieren digital Der CE-Routenplaner Strukturierte Produktentwicklung
Interpretation der Anforderungen Canisha Teubert Online-Handbücher für Technik, IT, Produktentwicklung Medizinprodukte planen, entwickeln, realisieren digital Der CE-Routenplaner Strukturierte Produktentwicklung
Erfahrungen der. DQS GmbH. bei der Zertifizierung von Medizinprodukteherstellern
 Erfahrungen der DQS GmbH bei der Zertifizierung von Medizinprodukteherstellern 2004-11-24, Seite 1 Normensituation Medizinprodukte DIN EN ISO 9001:94 DIN EN ISO 9001:2000 DIN EN 46001/2:1996 DIN EN ISO
Erfahrungen der DQS GmbH bei der Zertifizierung von Medizinprodukteherstellern 2004-11-24, Seite 1 Normensituation Medizinprodukte DIN EN ISO 9001:94 DIN EN ISO 9001:2000 DIN EN 46001/2:1996 DIN EN ISO
Checkliste Technische Dokumentation Produktname
 Teil 1: Allgemeines Qualitätsmanagement Nr. Vorgelegte Dokumentation Anmerkung 1 Managementhandbuch 2 Spezielle Prozessanweisungen, Verfahrensanweisungen und Arbeitsanweisungen 3 Mitgeltende Formblätter
Teil 1: Allgemeines Qualitätsmanagement Nr. Vorgelegte Dokumentation Anmerkung 1 Managementhandbuch 2 Spezielle Prozessanweisungen, Verfahrensanweisungen und Arbeitsanweisungen 3 Mitgeltende Formblätter
MDR Medical Device Regulation
 MDR Medical Device Regulation Grundlagen und Fristen zur Umsetzung Foie 1 MDR: Die wichtigsten Fristen auf einen Blick 05.05.2017 MDR veröffentlicht 25.05.2017 MDR tritt in Kraft 26.05.2020 MDR muss angewendet
MDR Medical Device Regulation Grundlagen und Fristen zur Umsetzung Foie 1 MDR: Die wichtigsten Fristen auf einen Blick 05.05.2017 MDR veröffentlicht 25.05.2017 MDR tritt in Kraft 26.05.2020 MDR muss angewendet
(Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION
 INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION") 15.7.2009 Amtsblatt der n Union C 163/1 IV (Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie
15.7.2009 Amtsblatt der n Union C 163/1 IV (Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie
Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014
 Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014 martin.zauner@fh-linz.at Inhalt Regulatorischer Rahmen für Medizinprodukte (regulatory affairs) Medizinische
Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014 martin.zauner@fh-linz.at Inhalt Regulatorischer Rahmen für Medizinprodukte (regulatory affairs) Medizinische
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten
 Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Allgemeiner Leitfaden zu CE-Kennzeichnung
 Emser Straße 19 65195 Wiesbaden Dipl.-Ing. Christoph Spreuer Verfahrens- und Sicherheitsingenieur Tel.: 0611 / 59 99 33 Fax.: 0611 / 59 01 17 E-Mail: info@spreuer.com Web: www.spreuer.com Allgemeiner Leitfaden
Emser Straße 19 65195 Wiesbaden Dipl.-Ing. Christoph Spreuer Verfahrens- und Sicherheitsingenieur Tel.: 0611 / 59 99 33 Fax.: 0611 / 59 01 17 E-Mail: info@spreuer.com Web: www.spreuer.com Allgemeiner Leitfaden
Normrevision DIN EN ISO 9001:2015. Seite: 1
 Seite: 1 Qualitätsmanagement nach DIN EN ISO 9001 neu Inhalt: Zeitplan für Unternehmen und Organisationen Gegenüberstellung der Gliederung der Versionen 2008 und 2015 Die wichtigsten Neuerungen und deren
Seite: 1 Qualitätsmanagement nach DIN EN ISO 9001 neu Inhalt: Zeitplan für Unternehmen und Organisationen Gegenüberstellung der Gliederung der Versionen 2008 und 2015 Die wichtigsten Neuerungen und deren
Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG)
") Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) ProdHaftG Ausfertigungsdatum: 15.12.1989 Vollzitat: "Produkthaftungsgesetz vom 15. Dezember 1989 (BGBl. I S. 2198),
Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) ProdHaftG Ausfertigungsdatum: 15.12.1989 Vollzitat: "Produkthaftungsgesetz vom 15. Dezember 1989 (BGBl. I S. 2198),
EINFÜHRUNG UND UMSETZUNG
 Thema DIN EN ISO 9001:2000 EINFÜHRUNG UND UMSETZUNG 1 Agenda Allgemein 9000:2000 Das neue Normenkonzept Umsetzung 2 Allgemein 3 Allgemein Warum neue Normen? 4 Allgemein Warum neue Normen? Überprüfungszyklus
Thema DIN EN ISO 9001:2000 EINFÜHRUNG UND UMSETZUNG 1 Agenda Allgemein 9000:2000 Das neue Normenkonzept Umsetzung 2 Allgemein 3 Allgemein Warum neue Normen? 4 Allgemein Warum neue Normen? Überprüfungszyklus
Produktvorstellung. Seiler. Regelwerk Technische Dokumentation nach EWG 93/42
 Produktvorstellung Qualitätsmanagement Verlag Regelwerk Technische Dokumentation nach EWG 93/42 Seiler Dokumentationen Zielgruppe: Dieses Regelwerk ist für Inverkehrbringer von Medizinprodukten. Es beinhaltet
Produktvorstellung Qualitätsmanagement Verlag Regelwerk Technische Dokumentation nach EWG 93/42 Seiler Dokumentationen Zielgruppe: Dieses Regelwerk ist für Inverkehrbringer von Medizinprodukten. Es beinhaltet
Qualitätsmanagement Änderungen in der ISO 9001 Entwurf 2007
 Qualitätsmanagement Änderungen in der ISO 9001 Entwurf 2007 30. Januar 2008 München Ablauf der Revision 2008 Start Systematic Review 15.12.2003 Ermittlung von Kundenfeedback 01/2004 Justification study
Qualitätsmanagement Änderungen in der ISO 9001 Entwurf 2007 30. Januar 2008 München Ablauf der Revision 2008 Start Systematic Review 15.12.2003 Ermittlung von Kundenfeedback 01/2004 Justification study
(Veröffentlichung der Titel und der Bezugsnummern der harmonisierten Normen im Sinne der Harmonisierungsrechtsvorschriften
 C 389/22 DE Amtsblatt der Europäischen Union 17.11.2017 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten
C 389/22 DE Amtsblatt der Europäischen Union 17.11.2017 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten
Erfahrungen aus der Zertifizierung
 DGSV-Kongress 2010 08.10.2010, Fulda Erfahrungen aus der Zertifizierung von Qualitätsmanagement-Systemen nach DIN EN ISO 13485 für die Aufbereitung von Medizinprodukten Dr. Thomas Kießling Sachverständiger
DGSV-Kongress 2010 08.10.2010, Fulda Erfahrungen aus der Zertifizierung von Qualitätsmanagement-Systemen nach DIN EN ISO 13485 für die Aufbereitung von Medizinprodukten Dr. Thomas Kießling Sachverständiger
Gegenüberstellung von DIN EN ISO 9001:2015. und DIN EN ISO 9001:2008
 Gegenüberstellung von DIN EN ISO 9001:2015 und DIN EN ISO 9001:2008 DIN EN ISO 9001:2015 DIN EN ISO 9001:2008 4 Kontext der Organisation weitgehend neu, siehe aber auch 1 Anwendungsbereich 4.1 Verstehen
Gegenüberstellung von DIN EN ISO 9001:2015 und DIN EN ISO 9001:2008 DIN EN ISO 9001:2015 DIN EN ISO 9001:2008 4 Kontext der Organisation weitgehend neu, siehe aber auch 1 Anwendungsbereich 4.1 Verstehen
Block 3 Entwicklung Verifizierung / Validierung. Inhaltsverzeichnis
 Block 3 Entwicklung Verifizierung / Validierung Inhaltsverzeichnis 1 Änderungsnachweis... 2 2 Einleitung... 2 3 Definitionen und Abkürzungen... 2 4 Referenzen... 2 5 Entwicklung - EN 9100 Ergänzungen und
Block 3 Entwicklung Verifizierung / Validierung Inhaltsverzeichnis 1 Änderungsnachweis... 2 2 Einleitung... 2 3 Definitionen und Abkürzungen... 2 4 Referenzen... 2 5 Entwicklung - EN 9100 Ergänzungen und
Bedeutung der Norm EN ISO GCP für Medizinprodukte in der Praxis -
 Bedeutung der Norm EN ISO 14155 - GCP für Medizinprodukte in der Praxis - Dr. Violetta Zmuda AGES MEA, Institut Überwachung, Abteilung Klinische Prüfung violetta.zmuda@ages.at AGES-Gespräch Wien, 12. Oktober
Bedeutung der Norm EN ISO 14155 - GCP für Medizinprodukte in der Praxis - Dr. Violetta Zmuda AGES MEA, Institut Überwachung, Abteilung Klinische Prüfung violetta.zmuda@ages.at AGES-Gespräch Wien, 12. Oktober
Konformitätsbewertung 3.9 A 10
 Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 10 Besondere Fragestellungen bei der Zertifizierung von Qualitätsmanagementsystemen nach DIN EN ISO 13485 in Unternehmen der Orthopädie-,
Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 10 Besondere Fragestellungen bei der Zertifizierung von Qualitätsmanagementsystemen nach DIN EN ISO 13485 in Unternehmen der Orthopädie-,
Musterhandbuch Qualitätsmanagementplan
 Musterhandbuch Leseprobe DIN ISO 10005:2009 Konditionen: 14 Tage Rückgaberecht! Kein Abo! ISBN 978-3-942882-25-5 Auflage 1 Grundlagen... 2 Gültigkeit... 2 Ziel und Grund... 2 Abkürzungen... 2 Informationen...
Musterhandbuch Leseprobe DIN ISO 10005:2009 Konditionen: 14 Tage Rückgaberecht! Kein Abo! ISBN 978-3-942882-25-5 Auflage 1 Grundlagen... 2 Gültigkeit... 2 Ziel und Grund... 2 Abkürzungen... 2 Informationen...
Vorschlag für eine Allgemeine Produktdokumentation
 Vorschlag für eine Allgemeine Produktdokumentation 0.1 Deckblatt (Firma, Titel. lfd. Nr., Freigabevermerk, Unterschriften etc.) 0.2 Inhaltsverzeichnis (z. B. als Document Master File) 0.3 Allgemeine Hinweise
Vorschlag für eine Allgemeine Produktdokumentation 0.1 Deckblatt (Firma, Titel. lfd. Nr., Freigabevermerk, Unterschriften etc.) 0.2 Inhaltsverzeichnis (z. B. als Document Master File) 0.3 Allgemeine Hinweise
EUROPÄISCHE KOMMISSION
 10.7.2015 DE Amtsblatt der Europäischen Union C 226/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
10.7.2015 DE Amtsblatt der Europäischen Union C 226/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung)
") Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung) Vom 20. Dezember 2001,Bundesgesetzblatt I Nr. 72, S. 3854 22. Dezember 2001, geändert durch Bundesgesetzblatt I S. 4456 vom 4. Dezember
Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung) Vom 20. Dezember 2001,Bundesgesetzblatt I Nr. 72, S. 3854 22. Dezember 2001, geändert durch Bundesgesetzblatt I S. 4456 vom 4. Dezember
Umsetzungsfristen IVDR. EU Verordnung über in-vitro Diagnostika (IVDR )
") Umsetzungsfristen der EU Verordnung über in-vitro Diagnostika (IVDR ) SWISS MEDTECH / SVDI Konferenz MDR & IVDR Auswirkungen auf die Schweiz Bern, 23.März 2017 Dr. Dieter Schönwald TÜV SÜD Product Service
Umsetzungsfristen der EU Verordnung über in-vitro Diagnostika (IVDR ) SWISS MEDTECH / SVDI Konferenz MDR & IVDR Auswirkungen auf die Schweiz Bern, 23.März 2017 Dr. Dieter Schönwald TÜV SÜD Product Service
Gebrauchstauglichkeit von
 Gebrauchstauglichkeit von Medizinprodukten i d EN 60601-1-6:2006 und EN 62366:2008 Stefan Hofmann Tel.: +49 69 95427-262 Email: stefan.hofmann@dqs.de DQS Medizinprodukte GmbH Gebrauchstauglichkeit Gebrauchstauglichkeit
Gebrauchstauglichkeit von Medizinprodukten i d EN 60601-1-6:2006 und EN 62366:2008 Stefan Hofmann Tel.: +49 69 95427-262 Email: stefan.hofmann@dqs.de DQS Medizinprodukte GmbH Gebrauchstauglichkeit Gebrauchstauglichkeit
Konformitätsbewertung 3.9 A 3
 Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 3 Reihenfolge bei der Durchführung von Konformitätsbewertungsverfahren Artikel 11 der Richtlinie 93/42/EWG legt fest, welche Konformitätsbewertungsverfahren
Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 3 Reihenfolge bei der Durchführung von Konformitätsbewertungsverfahren Artikel 11 der Richtlinie 93/42/EWG legt fest, welche Konformitätsbewertungsverfahren
Gegenüberstellung von DIN EN ISO 9001:2015. und DIN EN ISO 9001:2008
 Gegenüberstellung von DIN EN ISO 9001:2015 und DIN EN ISO 9001:2008 IN EN ISO 9001:2015 DIN EN ISO 9001:2008 4 Kontext der Organisation weitgehend neu, siehe aber auch 1 Anwendungsbereich 4.1 Verstehen
Gegenüberstellung von DIN EN ISO 9001:2015 und DIN EN ISO 9001:2008 IN EN ISO 9001:2015 DIN EN ISO 9001:2008 4 Kontext der Organisation weitgehend neu, siehe aber auch 1 Anwendungsbereich 4.1 Verstehen
GSG 1.4 Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG)
") Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) Vom 15. Dezember 1989 (BGBl. I S. 2198) zuletzt geändert am 19. Juli 2002 (BGBl. I S. 2679) Der Bundestag hat das folgende
Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) Vom 15. Dezember 1989 (BGBl. I S. 2198) zuletzt geändert am 19. Juli 2002 (BGBl. I S. 2679) Der Bundestag hat das folgende
Qualitätsmanagement Verlag. Seiler. Dokumentationen. Vorlagen Technische Dokumentation. Gemäß VERORDNUNG (EU) 2017/745. Auflage 1
 2017/745. Auflage 1") Qualitätsmanagement Verlag Seiler Dokumentationen Vorlagen Technische Dokumentation Gemäß VERORDNUNG (EU) 2017/745 Auflage 1 Liste der Dokumente Produktakte Dokument Revision vom Ersteller/-in Verteiler
Qualitätsmanagement Verlag Seiler Dokumentationen Vorlagen Technische Dokumentation Gemäß VERORDNUNG (EU) 2017/745 Auflage 1 Liste der Dokumente Produktakte Dokument Revision vom Ersteller/-in Verteiler
Niederspannungsrichtlinie 2006/95/EG 1. GPSGV. Siemens AG Alle Rechte vorbehalten.
 Niederspannungsrichtlinie 2006/95/EG 1. GPSGV Hintergründe ganz allgemein Verbesserung / Beibehaltung des Sicherheitsniveaus Abbau von Handelshemmnissen. Gewährleistung des freien Warenverkehrs in der
Niederspannungsrichtlinie 2006/95/EG 1. GPSGV Hintergründe ganz allgemein Verbesserung / Beibehaltung des Sicherheitsniveaus Abbau von Handelshemmnissen. Gewährleistung des freien Warenverkehrs in der
DIN EN ISO 9001:2015 DIN EN ISO 1 Anwendungsbereich 1 Anwendungsbereich 1.1 Allgemeines 2 Normative Verweisungen 2 Normative Verweisungen Freie 3 Nutz
 DIN EN ISO 9001:2015 DIN EN ISO 1 Anwendungsbereich 1 Anwendungsbereich 1.1 Allgemeines 2 Normative Verweisungen 2 Normative Verweisungen Freie 3 Nutzung Begriffe kostenloser Tools 3 Begriffe und Experten-Links
DIN EN ISO 9001:2015 DIN EN ISO 1 Anwendungsbereich 1 Anwendungsbereich 1.1 Allgemeines 2 Normative Verweisungen 2 Normative Verweisungen Freie 3 Nutzung Begriffe kostenloser Tools 3 Begriffe und Experten-Links
In-vitro-Diagnostika (IVD)
") In-vitro-Diagnostika (IVD) Qualifikationen für die Medizinprodukteindustrie Seminare 2017 TÜV SÜD Akademie GmbH Fachliche Leitung Birgit Klusmeier E-Mail: akd.medizintechnik@tuev-sued.de Manager In-vitro-Diagnostika
In-vitro-Diagnostika (IVD) Qualifikationen für die Medizinprodukteindustrie Seminare 2017 TÜV SÜD Akademie GmbH Fachliche Leitung Birgit Klusmeier E-Mail: akd.medizintechnik@tuev-sued.de Manager In-vitro-Diagnostika
Die neue IVD-Verordnung (IVDR)
") Die neue IVD-Verordnung* (IVDR) Standort Tirol: In-vitro Diagnostika auf dem Prüfstand Innsbruck, 20.03.2018 Dr. Dieter Schönwald * = Verordnung (EU) 2017 / 746 Folie 1 Agenda - 1 Wichtige Änderungen und
Die neue IVD-Verordnung* (IVDR) Standort Tirol: In-vitro Diagnostika auf dem Prüfstand Innsbruck, 20.03.2018 Dr. Dieter Schönwald * = Verordnung (EU) 2017 / 746 Folie 1 Agenda - 1 Wichtige Änderungen und
3 Jahre Zertifizierungen der Aufbereitung
 ÖGSV-wfhss-Congress 2007, Baden bei Wien, 05.05.2007 3 Jahre Zertifizierungen der Aufbereitung auf Basis der deutschen RKI/BfArM-Empfehlung Ein Erfahrungsbericht Dr. Thomas Kießling Sachverständiger für
ÖGSV-wfhss-Congress 2007, Baden bei Wien, 05.05.2007 3 Jahre Zertifizierungen der Aufbereitung auf Basis der deutschen RKI/BfArM-Empfehlung Ein Erfahrungsbericht Dr. Thomas Kießling Sachverständiger für
Praktisches Vorgehen zur Erreichung der MDR Readiness SAQ Fachgruppe Medizinprodukte 14. Juni 2018, Olten
 Praktisches Vorgehen zur Erreichung der MDR Readiness SAQ Fachgruppe Medizinprodukte 14. Juni 2018, Olten André Sauter Manager Regulatory Affairs Agenda 1. Yspomed s Infusions- und Injektions-Systeme (Beispiele)
Praktisches Vorgehen zur Erreichung der MDR Readiness SAQ Fachgruppe Medizinprodukte 14. Juni 2018, Olten André Sauter Manager Regulatory Affairs Agenda 1. Yspomed s Infusions- und Injektions-Systeme (Beispiele)
Anforderungen an die Aufbereitung von Medizinprodukten Gesundheitssystem Pflichten Reinigung Desinfektion Sterilisation Haftung
 Positionspapier Anforderungen an die Aufbereitung von Medizinprodukten Pflichten der Hersteller und Pflichten der GesundheitssystemAufbereitun Pflichten Reinigung Desinfektion Sterilisation Haftung Hygiene
Positionspapier Anforderungen an die Aufbereitung von Medizinprodukten Pflichten der Hersteller und Pflichten der GesundheitssystemAufbereitun Pflichten Reinigung Desinfektion Sterilisation Haftung Hygiene
Dr. Berthold Schäfer Bundesverband Baustoffe Steine und Erden e.v.
 Dr. Berthold Schäfer Bundesverband Baustoffe Steine und Erden e.v. Bundesverband Baustoffe Steine und Erden e.v. Die neue Bauproduktenverordnung aus Sicht der Hersteller Dr.-Ing. Berthold Schäfer Übergeordnete
Dr. Berthold Schäfer Bundesverband Baustoffe Steine und Erden e.v. Bundesverband Baustoffe Steine und Erden e.v. Die neue Bauproduktenverordnung aus Sicht der Hersteller Dr.-Ing. Berthold Schäfer Übergeordnete
Gegenüberstellung von ISO 9001:2015 und ISO 9001:2008
 von ISO 9001:2015 und ISO 9001:2008 ISO 9001:2015 ISO 9001:2008 4 Kontext der 4 Qualitätsmanagementsystem 4.1 Verstehen der und ihres Kontextes 4 5.6 4.2 Verstehen der Erfordernisse und Erwartungen interessierter
von ISO 9001:2015 und ISO 9001:2008 ISO 9001:2015 ISO 9001:2008 4 Kontext der 4 Qualitätsmanagementsystem 4.1 Verstehen der und ihres Kontextes 4 5.6 4.2 Verstehen der Erfordernisse und Erwartungen interessierter
Gegenüberstellung DIN EN 9100:2018 DIN EN 9100:2010
 Gegenüberstellung DIN EN 9100:2018 DIN EN 9100:2010 DIN EN 9100:2018 DIN EN 9100:2010 4 Kontext der 4 Qualitätsmanagementsystem 4.1 Verstehen der und ihres Kontextes 4 5.6 4.2 Verstehen der Erfordernisse
Gegenüberstellung DIN EN 9100:2018 DIN EN 9100:2010 DIN EN 9100:2018 DIN EN 9100:2010 4 Kontext der 4 Qualitätsmanagementsystem 4.1 Verstehen der und ihres Kontextes 4 5.6 4.2 Verstehen der Erfordernisse
Gegenüberstellung der Normkapitel
 Zuordnungstabelle ISO 9001: 2015 ISO 9001:2008 ISO 9001:2015 ISO 9001:2008 4 Kontext der Organisation 4.0 Qualitätsmanagementsystem 4.1 Verstehen der Organisation und Ihres 4.0 Qualitätsmanagementsystem
Zuordnungstabelle ISO 9001: 2015 ISO 9001:2008 ISO 9001:2015 ISO 9001:2008 4 Kontext der Organisation 4.0 Qualitätsmanagementsystem 4.1 Verstehen der Organisation und Ihres 4.0 Qualitätsmanagementsystem
DIN EN (VDE ): EN 62304: A1:2015
: EN 62304: A1:2015") Inhalt Vorwort...2 Europäisches Vorwort zu A1...3 Einleitung...10 1 Anwendungsbereich...14 1.1 *Zweck...14 1.2 *Anwendungsgebiet...14 1.3 Beziehung zu anderen Normen...14 1.4 Einhaltung...14 2 *Normative
Inhalt Vorwort...2 Europäisches Vorwort zu A1...3 Einleitung...10 1 Anwendungsbereich...14 1.1 *Zweck...14 1.2 *Anwendungsgebiet...14 1.3 Beziehung zu anderen Normen...14 1.4 Einhaltung...14 2 *Normative
Vergleich der Anforderungen der RiliBÄK K 2008 mit denen der DIN EN ISO 15189
 Vergleich der der K 2008 mit denen der Petra Möller und Gerd Hafner Zentrum für Labormedizin und Mikrobiologie, Essen 5.1 Das Labor muss unter fachlich qualifizierter Leitung stehen???????? Es besteht
Vergleich der der K 2008 mit denen der Petra Möller und Gerd Hafner Zentrum für Labormedizin und Mikrobiologie, Essen 5.1 Das Labor muss unter fachlich qualifizierter Leitung stehen???????? Es besteht
Der rechtliche Hintergrund für Medizinprodukte mit Schwerpunkt auf die Evaluierung der Biokompatibilität. Jaqueline Schierhuber Bonn, 1.
 Der rechtliche Hintergrund für Medizinprodukte mit Schwerpunkt auf die Evaluierung der Biokompatibilität Jaqueline Schierhuber Bonn, 1. Februar 2014 Die Themen Umsetzung von Normen Beurteilung der Biokompatibilität
Der rechtliche Hintergrund für Medizinprodukte mit Schwerpunkt auf die Evaluierung der Biokompatibilität Jaqueline Schierhuber Bonn, 1. Februar 2014 Die Themen Umsetzung von Normen Beurteilung der Biokompatibilität
Wesentliche Änderung EN 9100:2016 / EN 9120: München/Hamburg Michael Rotzsche/ Wolfgang Bott
 Wesentliche Änderung EN 9100:2016 / EN 9120:2016 Wesentliche Änderung EN 9100:2016 / EN 9120:2016 15.-16.03.2017 München/Hamburg Michael Rotzsche/ Wolfgang Bott 4.3 Festlegung des Anwendungsbereiches des
Wesentliche Änderung EN 9100:2016 / EN 9120:2016 Wesentliche Änderung EN 9100:2016 / EN 9120:2016 15.-16.03.2017 München/Hamburg Michael Rotzsche/ Wolfgang Bott 4.3 Festlegung des Anwendungsbereiches des
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854)
 vom 20. Dezember 2001 (BGBl. I. S. 3854)") Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Die Sicht des Gesetzgebers
 Essen, 23. Februar 2005 Aufbereitung von Medizinprodukten die Verantwortung des Herstellers und des Anwenders Die Sicht des Gesetzgebers Bundesministerium für Gesundheit und Soziale Sicherung 1 Gliederung
Essen, 23. Februar 2005 Aufbereitung von Medizinprodukten die Verantwortung des Herstellers und des Anwenders Die Sicht des Gesetzgebers Bundesministerium für Gesundheit und Soziale Sicherung 1 Gliederung
MARKTÜBERWACHUNG. Abschlussbericht der Schwerpunktaktion: Aktive Medizinprodukte mit Messfunktion. Nichtinvasive Blutdruckmessgeräte
 MARKTÜBERWACHUNG Abschlussbericht der Schwerpunktaktion: Aktive Medizinprodukte mit Messfunktion Nichtinvasive Blutdruckmessgeräte Marktüberwachung Interner Abschlussbericht der Schwerpunktaktion Aktive
MARKTÜBERWACHUNG Abschlussbericht der Schwerpunktaktion: Aktive Medizinprodukte mit Messfunktion Nichtinvasive Blutdruckmessgeräte Marktüberwachung Interner Abschlussbericht der Schwerpunktaktion Aktive
Thema: Haftung nach dem Produkthaftungsgesetz
 Aufgabenblatt Thema: Haftung nach dem Produkthaftungsgesetz 1. Unterscheiden Sie die Arten von Herstellern gemäß Produkthaftungsgesetz. Endprodukthersteller derjenige, der das Endprodukt tatsächlich hergestellt
Aufgabenblatt Thema: Haftung nach dem Produkthaftungsgesetz 1. Unterscheiden Sie die Arten von Herstellern gemäß Produkthaftungsgesetz. Endprodukthersteller derjenige, der das Endprodukt tatsächlich hergestellt
Qualitätssicherung aus Sicht der Industrie
 Qualitätssicherung aus Sicht der Industrie Essen, 01. Dezember 2005 präsentiert von Jürgen Kief PPBCE / Abteilung Technik Qualität (lat.: qualitas = Beschaffenheit, Eigenschaft) Qualität Ist ein Terminus
Qualitätssicherung aus Sicht der Industrie Essen, 01. Dezember 2005 präsentiert von Jürgen Kief PPBCE / Abteilung Technik Qualität (lat.: qualitas = Beschaffenheit, Eigenschaft) Qualität Ist ein Terminus
Die neue ATEX-Richtlinie 2014/34/EU Hans Christian Simanski
 Die neue ATEX-Richtlinie 2014/34/EU Hans Christian Simanski Die neue ATEX-Richtlinie 2014/34/EU BTEX ;-) Weshalb der Wechsel? Übergang zur neuen Richtlinie Was bleibt gleich? Was ändert sich? Hersteller
Die neue ATEX-Richtlinie 2014/34/EU Hans Christian Simanski Die neue ATEX-Richtlinie 2014/34/EU BTEX ;-) Weshalb der Wechsel? Übergang zur neuen Richtlinie Was bleibt gleich? Was ändert sich? Hersteller
Medizinproduktegesetz (MPG)
") Einführung ins Medizinproduktegesetz (MPG) Prof. Dr. Christian Fegeler Zielsetzung und Rechtlicher Rahmen Medical Device Direktive (MDD) Richtlinie i 93/42/EWG sowie Richtlinien 90/385/EWG aktive implantierbare
Einführung ins Medizinproduktegesetz (MPG) Prof. Dr. Christian Fegeler Zielsetzung und Rechtlicher Rahmen Medical Device Direktive (MDD) Richtlinie i 93/42/EWG sowie Richtlinien 90/385/EWG aktive implantierbare
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Anlage zur Akkreditierungsurkunde D-ZM nach DIN EN ISO/IEC :2015
 Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-17591-01-00 nach DIN EN ISO/IEC 17021-1:2015 Gültigkeitsdauer: 14.08.2017 bis 26.05.2020 Ausstellungsdatum: 14.08.2017 Urkundeninhaber:
Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-17591-01-00 nach DIN EN ISO/IEC 17021-1:2015 Gültigkeitsdauer: 14.08.2017 bis 26.05.2020 Ausstellungsdatum: 14.08.2017 Urkundeninhaber:
1 Haftung. 2 Produkt. 3 Fehler. (1) Ein Produkt hat einen Fehler, wenn es nicht die Sicherheit bietet, die unter Berücksichtigung aller Umstände,
 Ein Produkt hat einen Fehler, wenn es nicht die Sicherheit bietet, die unter Berücksichtigung aller Umstände,") Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) Vom 15. Dezember 1989 (BGBl. I S. 2198) zuletzt geändert durch Artikel 5 des Gesetzes vom 17. Juli 2017 (BGBl. I Nr.
Gesetz über die Haftung für fehlerhafte Produkte (Produkthaftungsgesetz - ProdHaftG) Vom 15. Dezember 1989 (BGBl. I S. 2198) zuletzt geändert durch Artikel 5 des Gesetzes vom 17. Juli 2017 (BGBl. I Nr.
CE-Kennzeichnung und EG-Konformität in der Praxis Überblick und Grundlagen
 CE-Kennzeichnung und EG-Konformität in der Praxis Überblick und Grundlagen B e g r i f f s e r k l ä r u n g Was ist EG-Konformität / CE-Kennzeichnung? CE = Communauté Européenne (Europäische Wirtschaftsgemeinschaft)
CE-Kennzeichnung und EG-Konformität in der Praxis Überblick und Grundlagen B e g r i f f s e r k l ä r u n g Was ist EG-Konformität / CE-Kennzeichnung? CE = Communauté Européenne (Europäische Wirtschaftsgemeinschaft)
Formblatt FB 001 Medizinprodukte: Überwachungsprotokoll zur VAW02_001 Überwachung des erstmaligen Inverkehrbringens von Medizinprodukten
 Sachbearbeitung: Datum der Überwachung: Überwachungsprotokoll Name und Anschrift des Verantwortlichen für das erstmalige Inverkehrbringen Hersteller Bevollmächtigter Einführer Name Straße PLZ, Ort Tel.:
Sachbearbeitung: Datum der Überwachung: Überwachungsprotokoll Name und Anschrift des Verantwortlichen für das erstmalige Inverkehrbringen Hersteller Bevollmächtigter Einführer Name Straße PLZ, Ort Tel.:
Legende: Keine Aktion erforderlich! Anpassung erforderlich Neue Anforderung
 Legende: Keine Aktion erforderlich! Anpassung erforderlich Neue Anforderung Abschnitte der ISO 9001:2008 Abschnitte der ISO 9001:2015 Delta- Anforderungen, Bemerkungen 4 Qualitätsmanagementsystem 4.4 QMS
Legende: Keine Aktion erforderlich! Anpassung erforderlich Neue Anforderung Abschnitte der ISO 9001:2008 Abschnitte der ISO 9001:2015 Delta- Anforderungen, Bemerkungen 4 Qualitätsmanagementsystem 4.4 QMS
wir müssen reden. Über Qualität!
 wir müssen reden. Über Qualität! "Made in Quality - Made for Success" 1 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack 1 Warum Auditors Liebling? Es ist eine muss Forderung jeder Systemnorm!
wir müssen reden. Über Qualität! "Made in Quality - Made for Success" 1 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack 1 Warum Auditors Liebling? Es ist eine muss Forderung jeder Systemnorm!
AMAH Aufbereitung in der Endoskopie
 AMAH Aufbereitung in der Endoskopie Risikobewertung von flexiblen Endoskopen und Folgerungen für die Aufbereitung Christian Roth Olympus Deutschland GmbH OLYMPUS Deutschland GmbH, Medical Systems Hannover,
AMAH Aufbereitung in der Endoskopie Risikobewertung von flexiblen Endoskopen und Folgerungen für die Aufbereitung Christian Roth Olympus Deutschland GmbH OLYMPUS Deutschland GmbH, Medical Systems Hannover,
4.3 Planung (Auszug ISO 14001:2004+Korr 2009) Die Organisation muss (ein) Verfahren einführen, verwirklichen und aufrechterhalten,
 Die Organisation muss (ein) Verfahren einführen, verwirklichen und aufrechterhalten,") 4.3 Planung (Auszug ISO 14001:2004+Korr 2009) 4.3.1 Umweltaspekte Die Organisation muss (ein) Verfahren einführen, verwirklichen und aufrechterhalten, a) um jene Umweltaspekte ihrer Tätigkeiten, Produkte
4.3 Planung (Auszug ISO 14001:2004+Korr 2009) 4.3.1 Umweltaspekte Die Organisation muss (ein) Verfahren einführen, verwirklichen und aufrechterhalten, a) um jene Umweltaspekte ihrer Tätigkeiten, Produkte
Dokumentenübersicht Revision vom Ersteller/-in Verteiler Grund der letzten Änderung
 Handbuch Kapitel 1 und 2 Anwendungsbereich & Normative Verweise 0 BdoL BdoL Kapitel 3 Begriffe Abkürzungen 0 BdoL BdoL Kapitel 4 Qualitätsmanagementsystem 0 BdoL BdoL Kapitel 5 Verantwortung der Leitung
Handbuch Kapitel 1 und 2 Anwendungsbereich & Normative Verweise 0 BdoL BdoL Kapitel 3 Begriffe Abkürzungen 0 BdoL BdoL Kapitel 4 Qualitätsmanagementsystem 0 BdoL BdoL Kapitel 5 Verantwortung der Leitung
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
DGSV-Kongress Dom zu Fulda
 DGSV-Kongress 2009 Dom zu Fulda 1 Die Rolle der Zertifizierung im Rahmen der Aufbereitung Dr. Undine Soltau Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten DGSV-Kongress
DGSV-Kongress 2009 Dom zu Fulda 1 Die Rolle der Zertifizierung im Rahmen der Aufbereitung Dr. Undine Soltau Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten DGSV-Kongress
Neue Regeln bei der Marktüberwachung. TÜV SÜD Product Service
 Neue Regeln bei der Marktüberwachung Wie ändert sich die Audit-Praxis? i Hans-Heiner Junker TÜV SÜD Product Service Folie 1 Bisherige Aufgaben & Kompetenzen Zukünftige Aufgaben & Kompetenzen Was ändert
Neue Regeln bei der Marktüberwachung Wie ändert sich die Audit-Praxis? i Hans-Heiner Junker TÜV SÜD Product Service Folie 1 Bisherige Aufgaben & Kompetenzen Zukünftige Aufgaben & Kompetenzen Was ändert
Die Öko-Design-Richtlinie als Handlungsrahmen für umweltverträgliche Medizinprodukte
 Die Öko-Design-Richtlinie als Handlungsrahmen für umweltverträgliche Medizinprodukte Vortrag im Rahmen der Veranstaltung Integrierte Produktpolitik in der Gesundheitswirtschaft 09. Februar 2005 Philips
Die Öko-Design-Richtlinie als Handlungsrahmen für umweltverträgliche Medizinprodukte Vortrag im Rahmen der Veranstaltung Integrierte Produktpolitik in der Gesundheitswirtschaft 09. Februar 2005 Philips
Kuratorium für Waldarbeit und Forsttechnik e.v. Die neue Maschinenrichtlinie 2006/42 EG
 Kuratorium für Waldarbeit und Forsttechnik e.v. Die neue Maschinenrichtlinie 2006/42 EG Was ändert sich? Allgemeines Die Richtlinie 2006/42/EG tritt am 29.12.2009 in Kraft; alle ab diesem Zeitpunkt in
Kuratorium für Waldarbeit und Forsttechnik e.v. Die neue Maschinenrichtlinie 2006/42 EG Was ändert sich? Allgemeines Die Richtlinie 2006/42/EG tritt am 29.12.2009 in Kraft; alle ab diesem Zeitpunkt in
1.2 Gesamtinhaltsverzeichnis
 Seite 1 1.2 1.2 1 Service und Verzeichnisse 1.1 Herausgeber- und Autorenverzeichnis 1.2 1.3 Stichwortverzeichnis 1.4 Zugang zum Internetportal 2 Rechtliche Grundlagen 2.1 Die neue Maschinenrichtlinie 2.1.1
Seite 1 1.2 1.2 1 Service und Verzeichnisse 1.1 Herausgeber- und Autorenverzeichnis 1.2 1.3 Stichwortverzeichnis 1.4 Zugang zum Internetportal 2 Rechtliche Grundlagen 2.1 Die neue Maschinenrichtlinie 2.1.1
Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG. Tamara Bauer
 Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG Tamara Bauer Inhalt Definitionen Richtlinien Kombinationsprodukte 2 Fallbeispiele: Medizinprodukt/Arzneimittel Medizinprodukt/Arzneimittel
Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG Tamara Bauer Inhalt Definitionen Richtlinien Kombinationsprodukte 2 Fallbeispiele: Medizinprodukt/Arzneimittel Medizinprodukt/Arzneimittel
EU-Konformitätserklärungen
 EU-Konformitätserklärungen Eine EU-Konformitätserklärung sagt aus, dass das betreffende Gerät/das elektrische Betriebsmittel/die Funkanlage den Anforderungen der zutreffenden Richtlinien entspricht. Es
EU-Konformitätserklärungen Eine EU-Konformitätserklärung sagt aus, dass das betreffende Gerät/das elektrische Betriebsmittel/die Funkanlage den Anforderungen der zutreffenden Richtlinien entspricht. Es
Medizinproduktehaftung
 Lehrstuhl Prof. Erwin Deutsch -Zentrum für Medizinrecht- Gosslerstr.19 37075 Göttingen E-Mail: edeutsc@gwdg.de Medizinproduktehaftung von Prof. Dr.iur., Dr. iur. med. h.c. mult., Dres. Erwin Deutsch vorgetragen
Lehrstuhl Prof. Erwin Deutsch -Zentrum für Medizinrecht- Gosslerstr.19 37075 Göttingen E-Mail: edeutsc@gwdg.de Medizinproduktehaftung von Prof. Dr.iur., Dr. iur. med. h.c. mult., Dres. Erwin Deutsch vorgetragen
Risikoanalyse (Schritt 1 bis 3) Risikobewertung (Schritt 4) Risikokontrolle (Schritt 5 bis 10) Gesamtrisikobewertung (Schritt 11)
 Risikobewertung (Schritt 4) Risikokontrolle (Schritt 5 bis 10) Gesamtrisikobewertung (Schritt 11)") Teil I. Teil II. Teil III. Teil IV. Teil V. Allgemeines - Gesetzliche Anforderungen - Risikomanagementprozess im Unternehmen - Verpflichtungserklärung der Leitung - Verantwortungen im Rahmen des Risikomanagements
Teil I. Teil II. Teil III. Teil IV. Teil V. Allgemeines - Gesetzliche Anforderungen - Risikomanagementprozess im Unternehmen - Verpflichtungserklärung der Leitung - Verantwortungen im Rahmen des Risikomanagements
Transition KaWa. Gleich geht es los, bestimmt. Auditcheckliste Jörg Roggensack TESTO 2014
 Transition KaWa Gleich geht es los, bestimmt. 1 wir müssen reden. Über Qualität! "Morgens um halb zehn in Ihrem Unternehmen" 2 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack Warum Auditors
Transition KaWa Gleich geht es los, bestimmt. 1 wir müssen reden. Über Qualität! "Morgens um halb zehn in Ihrem Unternehmen" 2 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack Warum Auditors
Agenda. Integration der CE-Prozesse im Unternehmen -Verfahren und Verantwortlichkeiten- Ziel des Vortrages. Vorteile der Integration
 Integration der CE-Prozesse im Unternehmen -Verfahren und Verantwortlichkeiten- 1 Agenda 1 2 3 4 5 Ziel des Vortrages Vorteile der Integration Hat sich etwas geändert in den Managementnormen? Praktische
Integration der CE-Prozesse im Unternehmen -Verfahren und Verantwortlichkeiten- 1 Agenda 1 2 3 4 5 Ziel des Vortrages Vorteile der Integration Hat sich etwas geändert in den Managementnormen? Praktische
Fragen der Produkthaftung im Hinblick auf den Betrieb unbemannter Schiffe Carina Lutter
 Fragen der Produkthaftung im Hinblick auf den Betrieb unbemannter Schiffe 29.06.2017 Carina Lutter Einfluss der Technik auf die Schiffsführung Sensoren (Kameras, Radar, Sonar, LiDAR)-> situation awareness
Fragen der Produkthaftung im Hinblick auf den Betrieb unbemannter Schiffe 29.06.2017 Carina Lutter Einfluss der Technik auf die Schiffsführung Sensoren (Kameras, Radar, Sonar, LiDAR)-> situation awareness
Validierung vs. Klinische Bewertung: Herausforderungen bei der praktischen Umsetzung. Validierung vs. Klinische Bewertung 1
 Validierung vs. Klinische Bewertung: Herausforderungen bei der praktischen Umsetzung Lukas Vogler, M.Sc. PROSYSTEM Software GmbH Dipl.-Ing. Sarah Panten PROSYSTEM Clinical Services GmbH Validierung vs.
Validierung vs. Klinische Bewertung: Herausforderungen bei der praktischen Umsetzung Lukas Vogler, M.Sc. PROSYSTEM Software GmbH Dipl.-Ing. Sarah Panten PROSYSTEM Clinical Services GmbH Validierung vs.
Risikomanagement als produktiver Beitrag zum Entwicklungsprozess
 Risikomanagement als produktiver Beitrag zum Entwicklungsprozess Alexander Fink MEDICA 2013 MEDICA 2013 Seite 1 Ziel des Vortrags ist die Verbindung zwischen Produkt(entwicklung) und Konformitätsbewertungsverfahren
Risikomanagement als produktiver Beitrag zum Entwicklungsprozess Alexander Fink MEDICA 2013 MEDICA 2013 Seite 1 Ziel des Vortrags ist die Verbindung zwischen Produkt(entwicklung) und Konformitätsbewertungsverfahren
Medizinproduktezulassung
 Medizinproduktezulassung Wie geht das heutzutage? Kurzes Streiflicht: Von der Idee zum zugelassen Produkt. Welche Tücken lauern am Wegesrand? Was bringt die Zukunft? Peter Hartung seleon GmbH, Leitung
Medizinproduktezulassung Wie geht das heutzutage? Kurzes Streiflicht: Von der Idee zum zugelassen Produkt. Welche Tücken lauern am Wegesrand? Was bringt die Zukunft? Peter Hartung seleon GmbH, Leitung
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), geändert durch Artikel 1 10 der Verordnung vom 4. Dezember 2002 (BGBl. I S. 4456) Auf Grund des
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), geändert durch Artikel 1 10 der Verordnung vom 4. Dezember 2002 (BGBl. I S. 4456) Auf Grund des
Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA Verordnung über Medizinprodukte Medizinprodukte-Verordnung
 juris Das Rechtsportal Gesamtes Gesetz Amtliche Abkürzung: MPV Ausfertigungsdatum: 20.12.2001 Gültig ab: 01.01.2002 Dokumenttyp: Rechtsverordnung Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA 7102-47-6
juris Das Rechtsportal Gesamtes Gesetz Amtliche Abkürzung: MPV Ausfertigungsdatum: 20.12.2001 Gültig ab: 01.01.2002 Dokumenttyp: Rechtsverordnung Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA 7102-47-6
Post Market Surveillance (PMS) - Erwartungen Die Bedeutung des PMCF im Rahmen
 - Erwartungen Die Bedeutung des PMCF im Rahmen") TÜV Süd Akademie Umsetzung der MDR/IVDR in der Praxis Post Market Surveillance (PMS) - Erwartungen Die Bedeutung des PMCF im Rahmen der Marktüberwachung Dr. Andrea Röthler Head of Project Management, Manager
TÜV Süd Akademie Umsetzung der MDR/IVDR in der Praxis Post Market Surveillance (PMS) - Erwartungen Die Bedeutung des PMCF im Rahmen der Marktüberwachung Dr. Andrea Röthler Head of Project Management, Manager
Protecting human health.
 IN EN ISO 17665 zur Validierung von Dampfsterilisationsprozessen NEUES von der Dampfsterilisation Gerald Göllner Leitung Technik der MMM Group Neues von der Dampfsterilisation EN 285 (neu in 2008/2009)
IN EN ISO 17665 zur Validierung von Dampfsterilisationsprozessen NEUES von der Dampfsterilisation Gerald Göllner Leitung Technik der MMM Group Neues von der Dampfsterilisation EN 285 (neu in 2008/2009)