Medizinprodukte. Merkblatt zur EU-Richtlinie 93/42/EWG (und 2007/47/EG)
|
|
|
- Kornelius Stein
- vor 7 Jahren
- Abrufe
Transkript
1 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 93/42/EWG (und 2007/47/EG)

2 Richtlinie über Sie stellen oder Zubehör her, handeln mit diesen Produkten oder importieren diese? Wissen Sie Bescheid über die rechtlichen Grundlagen? Können Sie nachweisen, dass Ihre Produkte den geltenden Sicherheitsbestimmungen entsprechen? Nein? Dann sollten Sie dieses Merkblatt aufmerksam lesen! Die EU-Richtlinie über muss in allen Mitgliedstaaten seit dem 14. Juni 1998 verbindlich angewendet werden. Rechtliche Grundlagen in der Europäischen Union (EU) in Deutschland Die Richtlinie 93/42/EWG des Rates vom 14. Juni 1993 über wurde im Amtsblatt der EG Nr. L 169 vom , S veröffentlicht und geändert durch: die Richtlinie 98/79/EG des Europäischen Parlaments und des Rates vom 27. Oktober 1998 über In-Vitro-Diagnostika (Amtsblatt Nr. L 331 vom 07/12/1998 S ); die Richtlinie 2000/70/EG des Europäischen Parlaments und des Rates vom 16. November 2000 zur Änderung der Richtlinie 93/42/EWG des Rates hinsichtlich n, die stabile Derivate aus menschlichem Blut oder Blutplasma enthalten (Amtsblatt Nr. L 313 vom 13/12/2000 S ); die Richtlinie 2001/104/EG vom 7. Dezember 2001 (Amtsblatt Nr. L 6 vom 10/01/2002) und die Richtlinie 2007/47/EG zur Angleichung der Rechtsvorschriften der Mitgliedsstaaten über aktive implantierbare medizinische Geräte und 93/42/EWG des Rates über sowie der Richtlinie über das Inverkehrbringen von Biozid-Produkten. Die wichtigsten deutschen Vorschriften zur Umsetzung der Medizinproduktrichtlinie sind: gesetz (MPG); verordnung (MPV); Verordnung über klinische Prüfungen von n (MPKPV) -Sicherheitsplanverordnung (MPSV); Informationssystem über (DIMDIV); -Betreiberverordnung (MPBetreibV); Verordnung über Vertriebswege für (MPVertrV); Verordnung über die Verschreibungspflicht von n (MPVerschrV). Die aktuellen deutschen Gesetzestexte sowie die EU-Richtlinien stehen unter der folgenden Webadresse zum Download zur Verfügung: 2
 in Deutschland Die Richtlinie 93/42/EWG des Rates vom 14. Juni 1993 über wurde im Amtsblatt der EG Nr. L 169 vom 12.7.1993, S.")
3 Geltungsbereich und wesentliche Änderungen Welche Produkte sind betroffen? Die EU-Richtlinie 93/42/EWG gilt für das Inverkehrbringen und die Inbetriebnahme von n und ihres Zubehörs. Seit dem gilt die neue Richtlinie 2007/47/EG. Sie ändert die Richtlinien 93/42/EWG und 90/385/EWG. müssen der neuen Richtlinie entsprechen, andernfalls dürfen sie nicht auf den europäischen Markt gebracht werden. Die wesentlichen Änderungen in der Richtlinie 2007/47/EG sind: Änderung der Klassifizierungsregeln und daher neue Einstufung einiger und u.u. anderes Konformitätsbewertungsverfahren; Änderung der grundlegenden Anforderungen, d.h. Neu-Bewertung der bereits CE gekennzeichneten Produkte wird erforderlich; Höhere Anforderungen an die technische Dokumentation; Hersteller, die unter ihrem Namen vertreiben und ihren Sitz außerhalb der Europäischen Union haben, dürfen die Produkte nur innerhalb der EU in Verkehr bringen, wenn sie einen einzigen Bevollmächtigten mit Sitz innerhalb der EU haben. Software gilt als Medizinprodukt, wenn sie spezifisch für einen oder mehrere der in der Definition von n genannten medizinischen Zwecke bestimmt ist; Verschärfte Anforderungen an klinische Daten, an deren Handhabung sowie an die Dokumentation, zum Teil auch für bestehende Produkte; richtlinie und Richtlinie über persönliche Schutzausrüstung 89/686/EWG müssen beide voll berücksichtigt werden; Medizingeräte, die auch als Maschine einzustufen sind, müssen zusätzlich die grundlegenden Anforderungen der EU-Maschinenrichtlinie 2006/42/EG erfüllen, wenn diese spezifischer sind als die der richtlinie. In den Anwendungsbereich der Richtlinie fallen alle einzeln oder miteinander verbunden verwendeten Instrumente, Apparate, Vorrichtungen, Software, Stoffe oder andere Gegenstände. Dazu gehört auch die vom Hersteller speziell zur Anwendung für diagnostische und/oder therapeutische Zwecke bestimmte und für ein einwandfreies Funktionieren des Medizinprodukts eingesetzte Software. Die Produkte sind vom Hersteller zur Anwendung am Menschen für folgende Zwecke vorgesehen: Erkennung, Verhütung, Überwachung, Behandlung oder Linderung von Krankheiten; Erkennung, Überwachung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen; Untersuchung, Ersatz oder Veränderung des anatomischen Aufbaus oder eines physiologischen Vorgangs; Empfängnisregelung. Die bestimmungsgemäße Hauptwirkung von n im oder am menschlichen Körper darf weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht werden. Ihre Wirkungsweise kann aber durch solche Mittel unterstützt werden. Außerdem ist die Richtlinie 93/42/EWG auf Zubehör von n anzuwenden. Zubehör sind Gegenstände, die selbst kein Produkt sind, sondern nach ihrer vom Hersteller speziell festgelegten Zweckbestimmung, zusammen mit einem Produkt zu verwenden sind. Dies sind z.b. Zusatzgeräte, EKG-Elektroden, Beatmungsschläuche oder Desinfektionsmittel für chirurgische Instrumente. 3

4 Auch einmalige Sonderanfertigungen für namentlich genannte Patienten und für klinische Prüfungen bestimmte Produkte fallen in den Anwendungsbereich der Richtlinie, tragen aber nicht die CE-Kennzeichnung. Produkte, die dazu bestimmt sind, ein Arzneimittel abzugeben, unterliegen ebenfalls dieser Richtlinie. Ausnahmen Wer ist davon betroffen? Welche Anforderungen enthält die Richtlinie? Diese Richtlinie gilt nicht für: Produkte für die In-vitro-Diagnose gemäß der Richtlinie 98/79/EG; aktive implantierbare medizinische Geräte gemäß der Richtlinie 90/385/EWG; Arzneimittel im Sinne der Richtlinie 2001/83/EG; kosmetische Mittel im Sinne der Richtlinie 76/768/EWG; menschliches Blut, Blutprodukte, Blutplasma oder Blutzellen menschlichen Ursprungs bzw. Produkte, die zum Zeitpunkt des Inverkehrbringens Blutprodukte, Blutplasma oder Blutzellen dieser Art enthalten; Transplantate, Gewebe oder Zellen menschlichen Ursprungs noch für Produkte, die Gewebe oder Zellen menschlichen Ursprungs enthalten oder aus solchen Geweben oder Zellen gewonnen wurden; Transplantate, Gewebe oder Zellen tierischen Ursprungs, es sei denn, ein Produkt wird unter Verwendung von abgetötetem tierischen Gewebe oder von abgetöteten Erzeugnissen hergestellt, die aus tierischem Gewebe gewonnen wurden. Von den Bestimmungen der Richtlinie betroffen sind: Der Hersteller oder der Bevollmächtigte des Herstellers in der EU und der Importeur oder die Person, die für das Inverkehrbringen des Produktes auf dem gemeinsamen Markt verantwortlich ist. Auch der Betreiber und der Anwender sind insofern betroffen, dass sie nur mit CE-Kennzeichnung in Betrieb nehmen dürfen. Grundsätzlich dürfen die Produkte nur in den Verkehr gebracht und in Betrieb genommen werden, wenn sie die Sicherheit und die Gesundheit der Patienten, der Anwender und gegebenenfalls Dritter bei sachgemäßer Installation, Instandhaltung und zweckgemäßer Verwendung nicht gefährden. Die Produkte müssen die grundlegenden Anforderungen gemäß Anhang I der Richtlinie 93/42/EG erfüllen, die auf sie unter Berücksichtigung ihrer Zweckbestimmung anwendbar sind. Diese umfassen neben allgemeinen Anforderungen vor allem Anforderungen an die Auslegung und Konstruktion des Produktes im Hinblick auf: Chemische, physikalische und biologische Eigenschaften der eingesetzten Werkstoffe (Toxizität, Verträglichkeit,...); Infektion und mikrobielle Kontamination (Verarbeitung, Verpackung,...); Eigenschaften im Hinblick auf die Konstruktion und die Umgebungsbedingungen (Minimierung von Risiken); Messfunktion (Genauigkeit, Anzeigeeinrichtungen,...); Schutz vor Strahlungen (beabsichtigte bzw. unbeabsichtigte Strahlung, ionisierende Strahlung,...); Produkte mit externer oder interner Energiequelle (Schutz vor elektrischen, mechanischen, thermischen Risiken,...); Bereitstellung von Informationen durch den Hersteller (Kennzeichnungen, Gebrauchsanweisung,...); Ermittlung von klinischen Daten. 4

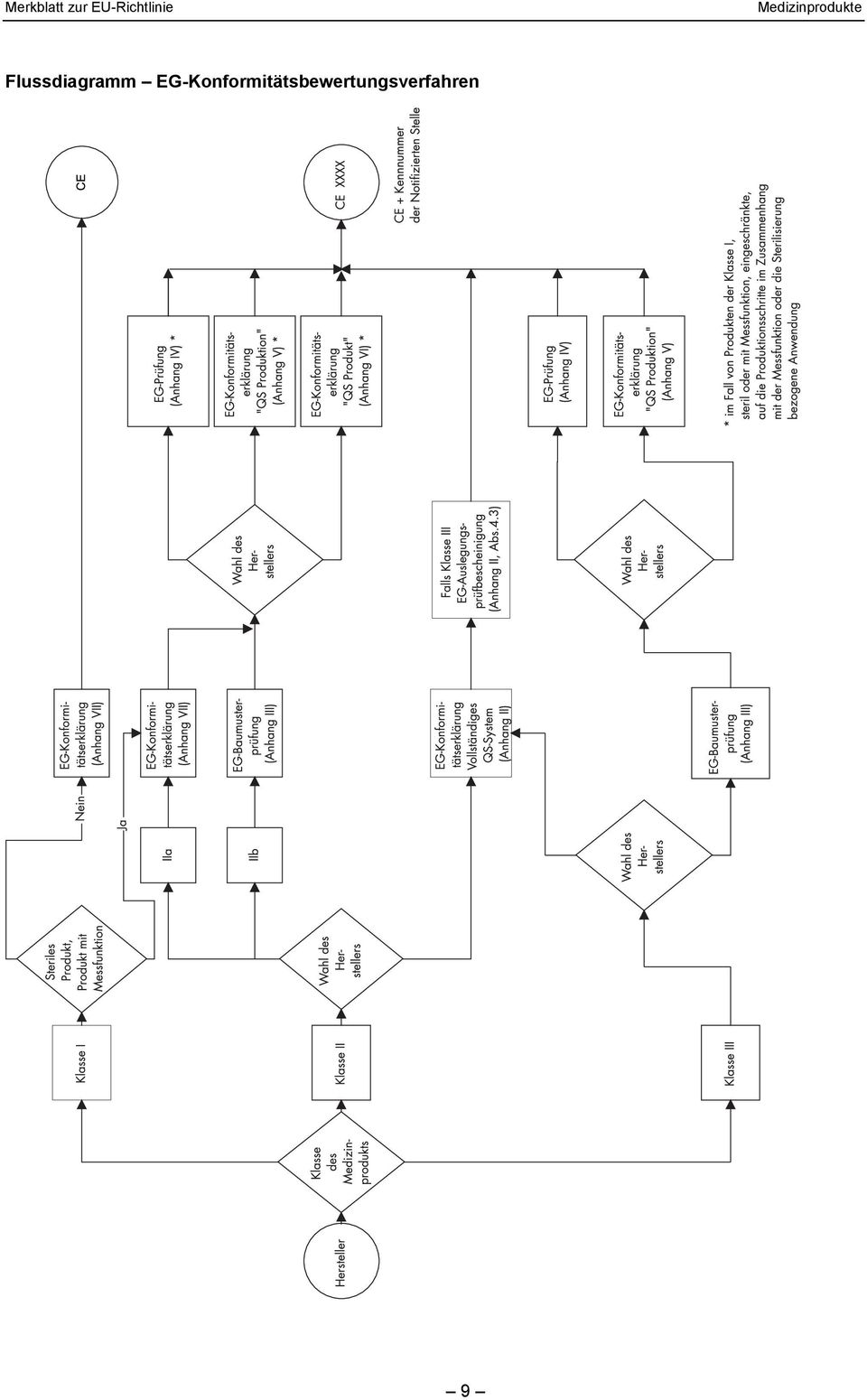
5 Klinische Bewertung Die Eignung von n für den vorgesehenen Verwendungszweck ist gemäß Anhang X der Richtlinie durch eine klinische Bewertung anhand von klinischen Daten zu belegen, soweit nicht in begründeten Ausnahmefällen andere Daten ausreichend sind. Die klinische Bewertung schließt die Beurteilung unerwünschter Wirkungen sowie die Annehmbarkeit des in den Grundlegenden Anforderungen der Richtlinien 93/42/EWG genannten Nutzen-/Risiko-Verhältnisses ein. Die klinische Bewertung muss gemäß eines definierten, methodisch einwandfreien Verfahrens erfolgen und gegebenenfalls einschlägige harmonisierte Normen berücksichtigen. Klassifizierung der Produkte werden entsprechend ihres Gefährdungspotenzials nach den in Anhang IX der Richtlinie festgelegten Klassifizierungskriterien in die Klassen I (niedrig), IIa, IIb und III (hoch) eingestuft. Die Klassifizierungsregeln basieren auf der Verletzbarkeit des menschlichen Körpers und berücksichtigen die potenziellen Risiken im Zusammenhang mit der technischen Auslegung und Herstellung der Produkte. Um die Klassifizierung zu erleichtern, hat die EU folgende Leitlinien herausgegeben: MEDDEV-Guidelines for the classification of medical devices. Diese sind (nur in englischer Sprache) auf folgender Webseite zu finden: Für die unterschiedlichen Produktklassen kommen unterschiedliche Verfahren zur Konformitätsbewertung zur Anwendung (siehe Flussdiagramm EG-Konformitätsbewertungsverfahren). Verfahren zur Konformitätsbewertung: Produkte der Klasse I Bei n der Klasse I, die nur geringe Gefahren beinhalten, wie Brillenfassungen oder Gehhilfen, kann der Hersteller oder sein Bevollmächtigter in eigener Verantwortung sicherstellen und erklären, dass die Produkte den einschlägigen Bestimmungen der richtlinie entsprechen. Er erstellt die technische Dokumentation und kennzeichnet die Produkte. Es handelt sich hierbei um das Verfahren der EG-Konformitätserklärung nach Anhang VII der Richtlinie. Für Produkte der Klasse I mit Messfunktion oder für sterile Produkte der Klasse I ist zusätzlich ein Verfahren nach Anhang IV, V oder VI anzuwenden, jeweils beschränkt auf die speziellen Anforderungen. Verfahren zur Konformitätsbewertung Produkte der Klasse IIa Für Produkte der Klasse IIa, z.b. für Hörgeräte oder EKG-Geräte muss in Verbindung mit dem o.g. Verfahren der EG-Konformitätserklärung wahlweise zusätzlich eines der folgenden Verfahren angewendet werden: Verfahren der EG-Prüfung (Produktprüfung) gemäß Anhang IV (eingeschränkt): Am Beginn steht der Nachweis, dass das Produkt den grundlegenden Anforderungen nach Anhang I der Richtlinie entspricht. Des Weiteren trifft der Hersteller alle erforderlichen Maßnahmen, um im Fertigungsprozess die Übereinstimmung aller Produkte einer Serie mit den Anforderungen der Richtlinie sicherzustellen. Eine Notifizierte Stelle überprüft die Konformität der Produkte mit der technischen Dokumentation nach Anhang VII, Abschnitt 3. Verfahren der EG-Konformitätserklärung (Qualitätssicherung Produktion) gemäß Anhang V (eingeschränkt): Auch in diesem Fall ist zunächst der Nachweis erforderlich, dass das Produkt den grundlegenden Anforderungen nach Anhang I entspricht. Der Hersteller wendet in der Produktion und der Endkontrolle ein genehmigtes Qualitätssicherungssystem an, das von einer Notifizierten Stelle auditiert und überwacht wird. Die Basis dafür kann ein QM-System nach 5

6 DIN EN ISO sein. Durch das genehmigte QS-System gewährleistet der Hersteller, dass die Produkte im Einklang mit der technischen Dokumentation nach Anhang VII hergestellt werden. Verfahren der EG-Konformitätserklärung (Qualitätssicherung Produkt) gemäß Anhang VI (eingeschränkt): Neben dem Nachweis der Konformität des Produkts mit den grundlegenden Anforderungen nach Anhang I ist hier ein genehmigtes Qualitätssicherungssystem für die Endkontrolle des Produkts erforderlich, das ebenfalls von einer Notifizierten Stelle auditiert und überwacht wird. Die Basis hierfür kann ein QM-System nach DIN EN ISO bilden. Mit dem genehmigten QS-System gewährleistet der Hersteller, dass die Produkte im Einklang mit der technischen Dokumentation nach Anhang VII hergestellt werden. Verfahren der EG-Konformitätserklärung (vollständiges Qualitätssicherungssystem) gemäß Anhang II (eingeschränkt): Neben dem Nachweis der Konformität des Produktes mit den grundlegenden Anforderungen der Richtlinie nach Anhang I ist hier ein genehmigtes Qualitätssicherungssystem für die Auslegung, die Fertigung und die Endkontrolle des Produktes erforderlich, das von einer Notifizierten Stelle auditiert und überwacht wird. Die Basis hierfür kann ein QM-System nach DIN EN ISO bilden. Produkte der Klasse IIb Produkte der Klasse III Für Produkte der Klasse IIb, wie Beatmungsgeräte oder Infusionspumpen, muss der Hersteller nach eigener Wahl eines der folgenden Verfahren einhalten: Verfahren der EG-Konformitätserklärung (vollständiges Qualitätssicherungssystem) gemäß Anhang II (eingeschränkt): Der Hersteller wendet in diesem Fall ein genehmigtes Qualitätssicherungssystem für die Auslegung, die Fertigung und die Endkontrolle der Produkte an, das von einer Notifizierten Stelle auditiert und überwacht wird. Die Grundlage hierfür kann ein QM-System nach DIN EN ISO13485 bilden. Verfahren der EG-Baumusterprüfung gemäß Anhang III: Hier bescheinigt eine Notifizierte Stelle, dass ein repräsentatives Exemplar der Produktion (Baumuster), den Bestimmungen dieser Richtlinie entspricht. Die zugehörige Dokumentation wird geprüft und bewertet. Das Verfahren der EG-Baumusterprüfung muss immer in Verbindung mit den Verfahren: der EG-Prüfung oder der EG-Konformitätserklärung (Qualitätssicherung Produktion) oder der EG-Konformitätserklärung (Qualitätssicherung Produkt) angewendet werden (siehe oben). Für die besonders risikoreichen Produkte der Klasse III, z.b. inaktive Implantate oder Nahtmaterialien zur Anwendung am Herzen, muss der Hersteller alternativ eines der folgenden Verfahren einhalten: Verfahren der EG-Konformitätserklärung (vollständige Qualitätssicherung) gemäß Anhang II (siehe oben). Zusätzlich wird vor der Herstellung eine Prüfung der Auslegungsdokumentation zu einem Produkt durch eine Notifizierte Stelle gefordert. Diese stellt eine EG-Auslegungsprüfbescheinigung aus. oder 6

7 Verfahren zur EG-Baumusterprüfung in Verbindung entweder mit dem Verfahren: der EG-Prüfung gemäß Anh. IV oder der EG-Konformitätserklärung (Qualitätssicherung Produktion) gemäß Anh. V. Es wird dann von der Einhaltung der grundlegenden Anforderungen der Richtlinie ausgegangen, wenn das Produkt den einschlägigen harmonisierten Normen i.s. des Art. 5 der Richtlinie entspricht (Konformitätsvermutung). Der Normenverweis schließt auch die Monographie des Europäischen Arzneibuchs ein, insbesondere über chirurgisches Nahtmaterial sowie die Aspekte der Wechselwirkung zwischen Arzneimitteln und Materialien von Produkten, die Arzneimittel aufnehmen. Im Amtsblatt der Europäischen Gemeinschaften wurden bereits zahlreiche harmonisierte Normen im Sinne dieser Richtlinie veröffentlicht. Beispiele für harmonisierte Normen sind die Normenreihen: DIN EN 556ff: Sterilisation von n Anforderungen an, die als steril gekennzeichnet werden DIN EN ISO 10993ff: Biologische Beurteilung von n DIN EN 60601ff: Medizinische elektrische Geräte Eine vollständige Liste der gültigen harmonisierten Normen kann unter folgender Internetadresse abgerufen werden: harmonised-standards-legislation/list-references/medical-devices/ index_en.htm Fehlen harmonisierte Normen, können zur Präzisierung der grundlegenden Anforderungen auch nationale Normen herangezogen werden. Die Anwendung von Normen ist grundsätzlich freiwillig. Was ist zu tun? Die Anbringung der CE-Kennzeichnung auf jedem Medizinprodukt ist Pflicht (mit Ausnahme von Sonderanfertigungen und von Produkten, die für die klinische Prüfung vorgesehen sind). Voraussetzung dafür ist ein vorher durchgeführtes Konformitätsbewertungsverfahren, in dem die Übereinstimmung des Produkts mit den grundlegenden Anforderungen der Richtlinie überprüft wird. Dabei muss das für die jeweilige Produktklasse vorgeschriebene Verfahren eingehalten werden. Es spielt in diesem Zusammenhang keine Rolle, ob das Gerät in der EU hergestellt oder aus Ländern außerhalb der EU importiert wird. Die Mitwirkung Notifizierter Stellen ist bei allen n vorgesehen. Ausgenommen sind Produkte der Klasse I, entweder ohne Messfunktion oder unsterile Produkte. Unterlagen, technische Dokumentation Die technische Dokumentation muss eine Bewertung der Konformität des Produktes mit den Anforderungen der Richtlinie ermöglichen. Sie muss insbesondere enthalten: eine allgemeine Beschreibung des Produktes, einschließlich der geplanten Varianten und seiner Zweckbestimmungen; Konstruktions- und Fertigungszeichnungen sowie Pläne von Bauteilen, Baugruppen, Schaltungen usw.; Beschreibungen und Erläuterungen der Zeichnungen und Pläne sowie der Funktionsweise des Produkts; 7

8 Ergebnisse der Risikoanalyse sowie eine Liste der ganz oder teilweise angewandten Normen; Beschreibung der Lösungen zur Einhaltung der grundlegenden Anforderungen dieser Richtlinie; sofern die Produkte in sterilem Zustand in den Verkehr gebracht werden, eine Beschreibung der angewandten Verfahren und den Validierungsbericht; Ergebnisse von Berechnungen, Prüfungen etc.; die präklinische Bewertung; die Prüfberichte und ggf. klinische Daten gemäß Anhang X; die Kennzeichnung sowie die Gebrauchsanweisung. Unterlagen, Technische Dokumentation Schriftliche Konformitätserklärung Hinzu kommt die Dokumentation des jeweils angewandten Qualitätssicherungssystems. Der vollständige Inhalt der technischen Unterlagen wird in den jeweiligen Anhängen der Richtlinie genannt. Die technischen Unterlagen müssen für den Zeitraum der Lebensdauer des Produktes zur Einsichtnahme durch die Überwachungsbehörde bereitgehalten werden, mindestens jedoch 10 Jahre lang nach der Herstellung des letzten Produktes. Bei Implantaten beträgt die Frist 15 Jahre. Der Hersteller oder sein in der Gemeinschaft niedergelassener Bevollmächtigter ist verpflichtet, im Rahmen eines nach der richtlinie vorgeschriebenen Konformitätsbewertungsverfahrens eine EG-Konformitätserklärung auszustellen, bevor das Produkt in Verkehr gebracht wird. Mit dieser schriftlichen Konformitätserklärung wird bestätigt, dass das in Verkehr gebrachte Produkt alle einschlägigen Sicherheitsanforderungen erfüllt. Es wird zudem üblicherweise beschrieben, welche anderen zutreffenden EG- Richtlinien eingehalten oder ob ggf. dafür geltende Übergangsregelungen in Anspruch genommen wurden. 8

9 Flussdiagramm EG-Konformitätsbewertungsverfahren 9
10 Zuständige Ministerien für die Marktüberwachung in Bayern Bayerisches Staatsministerium für Umwelt und Gesundheit Rosenkavalierplatz München Tel.: Fax: Notifizierte Stellen in Bayern TÜV Rheinland LGA Products GmbH Tillystraße Nürnberg Tel.: Fax: intercert@de.tuv.com TÜV SÜD Gruppe TÜV SÜD Product Service GmbH Ridlerstr München Tel.: Fax: info@tuev-sued.de Alle in der EU notifizierten Stellen sind in der NANDO-Datenbank abrufbar: Anbringen der CE-Kennzeichnung Mit Ausnahme von Sonderanfertigungen und Produkten, die für klinische Prüfungen bestimmt sind, müssen alle im Sinne dieser Richtlinie die CE-Kennzeichnung tragen. Die CE-Kennzeichnung muss, sofern dies durchführbar und zweckmäßig ist, vom Hersteller oder seinem Bevollmächtigten in der Europäischen Gemeinschaft auf dem Produkt direkt oder auf dem sterilen Verpackungsmaterial sowie auf der Gebrauchsanweisung angebracht sein. Wenn möglich, muss sie auch auf der Handelsverpackung angebracht sein. Der CE-Kennzeichnung muss die Kennnummer der ggf. eingeschalteten notifizierten Stelle hinzugefügt sein. Bei Produkten Klasse I entfällt diese Kennnummer. Die Mindesthöhe der CE-Kennzeichnung beträgt 5 mm. Die geometrischen Abmessungen müssen exakt eingehalten sein (siehe nebenstehende Kennzeichnung). XXXX (Kenn-Nummer der Notifizierten Stelle) Praktische Hinweise für Deutschland Registrierung des Inverkehrbringers und der (Anzeigepflicht): Jeder Hersteller, der erstmalig in Deutschland in Verkehr bringt, muss den zuständigen Behörden die Anschrift seines Firmensitzes und die Beschreibung der betreffenden Produkte mitteilen. Dies gilt auch für Firmen, die Systeme oder Behandlungseinheiten zusammensetzen. Die Anzeige muss vor der Aufnahme der Tätigkeit erfolgen. Nachträgliche Änderungen der Angaben sowie die Einstellung des Inverkehrbringens sind unverzüglich anzuzeigen. 10

11 In Bayern sind die Bezirksregierungen für die Bearbeitung der Registrierung (Anzeige) zuständig. Die Anzeige kann online bei DIMDI unter folgender Internet-Adresse erfolgen: Eine Liste der zuständigen Behörden ist unter folgender Internetadresse abrufbar: Sicherheitsbeauftragter für und berater: Jeder Verantwortliche für das Inverkehrbringen, der seinen Sitz in Deutschland hat, muss unverzüglich nach Aufnahme der Tätigkeit einen Sicherheitsbeauftragten für bestimmen. Diese Person muss über die erforderliche Sachkenntnis und Zuverlässigkeit verfügen (naturwissenschaftlicher, medizinischer oder technischer Hochschulabschluss oder andere Ausbildung, die sie befähigt, die Aufgaben zu bewältigen, 2 Jahre Berufserfahrung). Die Aufgaben eines Sicherheitsbeauftragten bestehen in der Sammlung und Bewertung von Meldungen über Risiken der sowie in der Koordination der notwendigen Gegenmaßnahmen. Er ist auch für die Erfüllung von Anzeigepflichten verantwortlich, so weit sie risiken betreffen. Der Verantwortliche muss den Sicherheitsbeauftragten bei der zuständigen Behörde melden und jeden Personenwechsel anzeigen. Wer berufsmäßig Fachkreise fachlich informiert oder in die sachgerechte Handhabung der einweist wird als berater bezeichnet. Er darf diese Tätigkeit nur ausüben, wenn er die für die jeweiligen erforderliche Sachkenntnis und Erfahrung für die Information und, soweit erforderlich, für die Einweisung in die Handhabung der jeweiligen besitzt. Dies gilt auch für die fernmündliche Information. Die Sachkenntnis besitzt, wer eine Ausbildung in einem naturwissenschaftlichen, medizinischen oder technischen Beruf erfolgreich abgeschlossen hat und auf die jeweiligen bezogen geschult worden ist oder durch eine mindestens einjährige Tätigkeit, die in begründeten Fällen auch kürzer sein kann, Erfahrungen in der Information über die jeweiligen und, soweit erforderlich, in der Einweisung in deren Handhabung erworben hat. Meldung von Vorkommnissen und Rückrufen (Anzeigepflicht): Um Risiken bei n zu erfassen, zu bewerten und abzuwehren, müssen Vorkommnisse und Rückrufe entsprechend der - Sicherheitsplanverordnung MPSV der zuständigen Bundesoberbehörde gemeldet werden. Dies ist das Bundesinstitut für Arzneimittel und (BfArM) in Bonn. Dazu sind die vorgesehenen Formblätter zu verwenden. Diese stehen auf der Homepage von DIMDI zum Download bereit Weitere Informationen Deutsches Institut für Medizinische Dokumentation und Information (DIMDI) Internet: Bundesinstitut für Arzneimittel und Internet: Bayerisches Landesamt für Arbeitsschutz Internet: 11

12 Die Mitglieder des Arbeitskreises Europäische Normung und Qualitätssicherung, die Notifizierten Stellen und die Bundesnetzagentur mit ihren Außenstellen stehen den Herstellern unterstützend zur Seite. Weitere Information und Beratung zur Produktkonformität erhalten Sie auch von den EU-Beratungsstellen des Enterprise-Europe-Network in Bayern Wichtig Für Betroffene ist es unerlässlich, über diese Kurzinformation hinaus die entsprechenden Richtlinien in der aktuellen Ausgabe eingehend zu studieren. Bezugsquellen für EU-Richtnien/ Gesetzestexte/Normen (Nur komplette Amtsblätter) Beuth Verlag Burggrafenstraße Berlin Tel.: Fax: info@beuth.de Internet: Bundesanzeiger Verlag Postfach Köln Tel.: Fax: Gesetzgebungsportal der EU (Download kostenlos) Deutsche Gesetze (Download kostenlos) TÜV Rheinland Consulting GmbH EU-Beratung Tillystraße Nürnberg Tel.: Fax: een@de.tuv.com Internet: vertrieb@bundesanzeiger.de Internet: Veröffentlichte Merkblätter zu EU-Richtlinien 2006/95/EG Sicherheit von elektrischen Betriebsmitteln 2009/48/EG Sicherheit von Spielzeug 305/2011/EU Verordnung über Bauprodukte (anzuwenden ab ) 2004/108/EG Elektromagnetische Verträglichkeit 89/686/EWG Persönliche Schutzausrüstungen 2009/23/EG Nichtselbsttätige Waagen 2009/142/EG Gasverbrauchseinrichtungen 92/42/EWG Wirkungsgrade von mit flüssigen oder gasförmigen Brennstoffen beschickten neuen Warmwasserheizkesseln 93/42/EWG u. 2007/47/EG 97/23/EG Sicherheit von Druckgeräten 2006/42/EG Sicherheit von Maschinen 1999/5/EG Funkanlagen und Telekommunikationsendeinrichtungen 2001/95/EG Allgemeine Produktsicherheit 2000/14/EG Umweltbelastende Geräuschemissionen von Geräten und Maschinen Anwendung von Normen im Rahmen der CE-Kennzeichnung CE-Kennzeichnung Überblick über die Rahmenregelungen 12
 Beuth Verlag Burggrafenstraße 6 10787 Berlin Tel.: 030 2601-2260 Fax: 030 2601-1260 E-Mail: info@beuth.")
13 Weitere Merkblätter und Leitfäden finden Sie auf der Internetseite des Bayerischen Staatsministeriums für Wirtschaft und Medien, Energie und Technologie, München. Das Merkblatt wurde im Auftrag des Bayerischen Staatsministeriums für Wirtschaft und Medien, Energie und Technologie in Gemeinschaftsarbeit von den Mitgliedern des Arbeitskreises Europäische Normung und Qualitätssicherung erstellt und abgestimmt. Die Druckschrift wurde mit großer Sorgfalt zusammengestellt. Gewähr für die Richtigkeit und Vollständigkeit des Inhalts kann dessen ungeachtet nicht übernommen werden. Mitglieder des Arbeitskreises Europäische Normung und Qualitätssicherung beim Bayerischen Staatsministerium für Wirtschaft und Medien, Energie und Technologie: Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Christoph Pfaff Herbert Jung München Tel.: Fax: Bayerisches Staatsministerium für Umwelt und Verbraucherschutz Martin Schinke Dr. Matthias Honnacker Rosenkavalierplatz München Tel.: Fax: martin.schinke@stmuv.bayern.de Bayerisches Staatsministerium des Innern, für Bau und Verkehr Gerd Ackermann Georg Feuchtgruber Franz-Josef-Strauß-Ring München Tel.: Fax: georg.feuchtgruber@stmi.bayern.de TÜV Rheinland Akademie GmbH Dr. Monika Bias Edwin Schmitt Tillystraße Nürnberg Tel.: Fax: monika.bias@de.tuv.com Bayerischer Industrie- und Handelskammertag (BIHK) Karen Tittel Balanstraße München Tel.: Fax: karen.tittel@muenchen.ihk.de Bayerischer Handwerkstag e.v. (BHT) Raik Hoffmann Max-Joseph-Straße München Tel.: Fax: raik.hoffmann@hwk-muenchen.de Landesverband Groß- und Außenhandel, Vertrieb und Dienstleistungen Bayern e. V. Dr. Wolfgang Bauer Max-Joseph-Straße München Tel.: Fax: info@lgad.de TÜV SÜD AG Konzernbereich für Akkreditierung, Zertifizierung und Normenwesen Christian Priller Monika Weigel-Hafner Westendstraße München Tel.: Fax: christian.priller@tuev-sued.de DIN Deutsches Institut für Normung e. V. Ausschuss Normenpraxis (ANP) im DIN Patricia Dind M. A. Am DIN-Platz, Burggrafenstraße Berlin Tel.: Fax: patricia.dind@din.de 13

14 Impressum Herausgeber: Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Prinzregentenstraße 28, München Tel.: , Fax: Internet: Stand: 01/2014 in Zusammenarbeit mit dem Arbeitskreis Europäische Normung und Qualitätssicherung 14

Guideline für Klasse I Hersteller Abgrenzung und Klassifizierung
 Guideline für Klasse I Hersteller Abgrenzung und Klassifizierung CMI Workshop Medizinprodukte Klasse I Vademecum für den Marktzugang 29.11.2011 Bundesministerium für Gesundheit Seite 1 Aufgabenbereiche
Guideline für Klasse I Hersteller Abgrenzung und Klassifizierung CMI Workshop Medizinprodukte Klasse I Vademecum für den Marktzugang 29.11.2011 Bundesministerium für Gesundheit Seite 1 Aufgabenbereiche
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie. Gasverbrauchseinrichtungen. Merkblatt zur EU-Richtlinie 2009/142/EG
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2009/142/EG Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln mit ihnen oder importieren
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2009/142/EG Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln mit ihnen oder importieren
Merkblatt zur EU-Richtlinie 88/378/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 88/378/EWG Stand: März 2005 Sie stellen Spielzeug her, handeln mit diesen Produkten oder
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 88/378/EWG Stand: März 2005 Sie stellen Spielzeug her, handeln mit diesen Produkten oder
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), geändert durch Artikel 1 10 der Verordnung vom 4. Dezember 2002 (BGBl. I S. 4456) Auf Grund des
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), geändert durch Artikel 1 10 der Verordnung vom 4. Dezember 2002 (BGBl. I S. 4456) Auf Grund des
Med-Info. Richtlinie 93/42/EWG über Medizinprodukte. TÜV SÜD Product Service GmbH. Internationale Fach-Informationen für die Medizinproduktebranche
 Med-Info Internationale Fach-Informationen für die Medizinproduktebranche Richtlinie 93/42/EWG über Medizinprodukte Praxisorientierte Zusammenfassung der wichtigsten Aspekte und Anforderungen der Richtlinie
Med-Info Internationale Fach-Informationen für die Medizinproduktebranche Richtlinie 93/42/EWG über Medizinprodukte Praxisorientierte Zusammenfassung der wichtigsten Aspekte und Anforderungen der Richtlinie
Gasverbrauchseinrichtungen. Merkblatt zur EU-Richtlinie 90/396/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 90/396/EWG Stand: März 2005 Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 90/396/EWG Stand: März 2005 Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln
Medizinprodukte. Merkblatt zur EU-Richtlinie 93/42/EWG (und 2007/47/EG)
") Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 93/42/EWG (und 2007/47/EG) Richtlinie über Sie stellen oder Zubehör her, handeln mit diesen
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 93/42/EWG (und 2007/47/EG) Richtlinie über Sie stellen oder Zubehör her, handeln mit diesen
Merkblatt zur EU-Richtlinie 73/23/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Sicherheit von elektrischen Betriebsmitteln Merkblatt zur EU-Richtlinie 73/23/EWG Stand: März 2005 Niederspannungsrichtlinie
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Sicherheit von elektrischen Betriebsmitteln Merkblatt zur EU-Richtlinie 73/23/EWG Stand: März 2005 Niederspannungsrichtlinie
Vorschlag für eine Allgemeine Produktdokumentation
 Vorschlag für eine Allgemeine Produktdokumentation 0.1 Deckblatt (Firma, Titel. lfd. Nr., Freigabevermerk, Unterschriften etc.) 0.2 Inhaltsverzeichnis (z. B. als Document Master File) 0.3 Allgemeine Hinweise
Vorschlag für eine Allgemeine Produktdokumentation 0.1 Deckblatt (Firma, Titel. lfd. Nr., Freigabevermerk, Unterschriften etc.) 0.2 Inhaltsverzeichnis (z. B. als Document Master File) 0.3 Allgemeine Hinweise
Inhaltsverzeichnis. Inhaltsverzeichnis. Inhaltsübersicht. Rechtliche Grundlagen des Medizinprodukterechts 1
 Inhaltsverzeichnis Vorwort Inhaltsübersicht III X Rechtliche Grundlagen des Medizinprodukterechts 1 Richtlinie über Medizinprodukte 1 Rechtliche Grundlagen in der Europäischen Union (EU) 1 Rechtliche Grundlagen
Inhaltsverzeichnis Vorwort Inhaltsübersicht III X Rechtliche Grundlagen des Medizinprodukterechts 1 Richtlinie über Medizinprodukte 1 Rechtliche Grundlagen in der Europäischen Union (EU) 1 Rechtliche Grundlagen
Hilfe im Gesetzesdschungel der Medizinprodukte
 Hilfe im Gesetzesdschungel der Medizinprodukte Praktische Anwendung des Leitfadens zum Umgang mit Medizinprodukten Dr. Karin Kaster DLRG OG St. Wendel e.v. Stellv. Vorsitzende karin.kaster@stwendel.dlrg.de
Hilfe im Gesetzesdschungel der Medizinprodukte Praktische Anwendung des Leitfadens zum Umgang mit Medizinprodukten Dr. Karin Kaster DLRG OG St. Wendel e.v. Stellv. Vorsitzende karin.kaster@stwendel.dlrg.de
Sicherheit von elektrischen Betriebsmitteln
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2006/95/EG Niederspannungsrichtlinie (Richtlinie über elektrische Betriebsmittel zur Verwendung
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2006/95/EG Niederspannungsrichtlinie (Richtlinie über elektrische Betriebsmittel zur Verwendung
Persönliche Schutzausrüstungen. Merkblatt zur EU-Richtlinie 89/686/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Stand: März 2005 Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Stand: März 2005 Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie. Gasverbrauchseinrichtungen. Merkblatt zur EU-Richtlinie 2009/142/EG
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2009/142/EG Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln mit ihnen oder importieren
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2009/142/EG Gasgeräterichtlinie (Richtlinie über ) Sie stellen her, handeln mit ihnen oder importieren
Formblatt FB 001 Medizinprodukte: Überwachungsprotokoll zur VAW02_001 Überwachung des erstmaligen Inverkehrbringens von Medizinprodukten
 Sachbearbeitung: Datum der Überwachung: Überwachungsprotokoll Name und Anschrift des Verantwortlichen für das erstmalige Inverkehrbringen Hersteller Bevollmächtigter Einführer Name Straße PLZ, Ort Tel.:
Sachbearbeitung: Datum der Überwachung: Überwachungsprotokoll Name und Anschrift des Verantwortlichen für das erstmalige Inverkehrbringen Hersteller Bevollmächtigter Einführer Name Straße PLZ, Ort Tel.:
9.1 Medizinproduktegesetz - MPG
 9.1 Medizinproduktegesetz - MPG Das Gesetz über den Verkehr mit Medizinprodukten (Medizinproduktegesetz - MPG) dient der Umsetzung der Richtlinien des Rates 90/385/EWG vom 20. Juni 1990 zur Angleichung
9.1 Medizinproduktegesetz - MPG Das Gesetz über den Verkehr mit Medizinprodukten (Medizinproduktegesetz - MPG) dient der Umsetzung der Richtlinien des Rates 90/385/EWG vom 20. Juni 1990 zur Angleichung
Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014
 Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014 martin.zauner@fh-linz.at Inhalt Regulatorischer Rahmen für Medizinprodukte (regulatory affairs) Medizinische
Wann wird Bildverarbeitungssoftware zum Medizinprodukt? 11. Linzer Forum Medizintechnik November 2014 martin.zauner@fh-linz.at Inhalt Regulatorischer Rahmen für Medizinprodukte (regulatory affairs) Medizinische
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854)
 vom 20. Dezember 2001 (BGBl. I. S. 3854)") Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz. SPECTARIS-Infoveranstaltung
 Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz SPECTARIS-Infoveranstaltung München, 10. Januar 2014 Was ist zu tun? - Warum ist meine Brille ein Medizinprodukt? - Welche Maßnahmen muss ich
Richtiges Brillen-Management nach dem Medizinprodukte-Gesetz SPECTARIS-Infoveranstaltung München, 10. Januar 2014 Was ist zu tun? - Warum ist meine Brille ein Medizinprodukt? - Welche Maßnahmen muss ich
Revision RL 93/42/EWG Aufgaben der Benannten Stelle
 RICHTLINIE 2007/47/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. September 2007 zur Änderung der Richtlinien 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über aktive
RICHTLINIE 2007/47/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. September 2007 zur Änderung der Richtlinien 90/385/EWG des Rates zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über aktive
Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung)
") Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung) Vom 20. Dezember 2001,Bundesgesetzblatt I Nr. 72, S. 3854 22. Dezember 2001, geändert durch Bundesgesetzblatt I S. 4456 vom 4. Dezember
Verordnung über Medizinprodukte - MPV (Medizinprodukte-Verordnung) Vom 20. Dezember 2001,Bundesgesetzblatt I Nr. 72, S. 3854 22. Dezember 2001, geändert durch Bundesgesetzblatt I S. 4456 vom 4. Dezember
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie. CE-Kennzeichnung. Merkblatt zur EU-Richtlinie 93/68/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 93/68/EWG Stand: März 2005 Was müssen Hersteller über die wissen? Anfang der 90er Jahre
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 93/68/EWG Stand: März 2005 Was müssen Hersteller über die wissen? Anfang der 90er Jahre
Umweltbelastende Geräuschemissionen von Geräten und Maschinen
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Umweltbelastende Geräuschemissionen von Geräten und Maschinen Merkblatt zur EU-Richtlinie 2000/14/EG und EU-Richtlinie 2005/88/EG
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Umweltbelastende Geräuschemissionen von Geräten und Maschinen Merkblatt zur EU-Richtlinie 2000/14/EG und EU-Richtlinie 2005/88/EG
Control 2010 Qualitätssicherung in der Medizintechnik
 Control 2010 Qualitätssicherung in der Geschäftsbereiche Consulting The Business Designers Investitionsunabhängige Beratung Management Consulting Compliance Consulting IS/CSV Consulting Engineering The
Control 2010 Qualitätssicherung in der Geschäftsbereiche Consulting The Business Designers Investitionsunabhängige Beratung Management Consulting Compliance Consulting IS/CSV Consulting Engineering The
Persönliche Schutzausrüstungen
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen (PSA), Schutzvorrichtungen,
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen (PSA), Schutzvorrichtungen,
Richtlinie über die Sicherheit von Spielzeug. Merkblatt zur EU-Richtlinie 2009/48/EG
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Richtlinie über die Sicherheit von Spielzeug Merkblatt zur EU-Richtlinie 2009/48/EG Sicherheit von Spielzeug Sie stellen
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Richtlinie über die Sicherheit von Spielzeug Merkblatt zur EU-Richtlinie 2009/48/EG Sicherheit von Spielzeug Sie stellen
Klinische Prüfung von Medizinprodukten in Deutschland
 Klinische Prüfung von Medizinprodukten in Deutschland Notwendigkeit und Anforderungen 9. DVMD-Fachtagung, 31.03.2006, Erlangen Anne Eichberger 3M 2006 1 Anne Eichberger 30.03.2006 Was ist ein Medizinprodukt?
Klinische Prüfung von Medizinprodukten in Deutschland Notwendigkeit und Anforderungen 9. DVMD-Fachtagung, 31.03.2006, Erlangen Anne Eichberger 3M 2006 1 Anne Eichberger 30.03.2006 Was ist ein Medizinprodukt?
Abschnitt 1 Anwendungsbereich und Allgemeine Anforderungen an die Konformitätsbewertung 1 Anwendungsbereich
 13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") 05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess
 CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess IHK Dialog Gesundheitswirtschaft: Medizintechnik in der Region Köln-Bonn 14. Februar 2017 2017 by qcmed GmbH, Dr. Carola
CE-Kennzeichnung von Medizinprodukten Handlungsempfehlungen für den Zulassungsprozess IHK Dialog Gesundheitswirtschaft: Medizintechnik in der Region Köln-Bonn 14. Februar 2017 2017 by qcmed GmbH, Dr. Carola
Richtlinie über die Sicherheit von Spielzeug. Merkblatt zur EU-Richtlinie 2009/48/EG
 Richtlinie über die Merkblatt zur EU-Richtlinie 2009/48/EG Sie stellen Spielzeug her, handeln mit diesen Produkten oder importieren sie? Wissen Sie Bescheid über die gesetzlichen Regelungen? Können Sie
Richtlinie über die Merkblatt zur EU-Richtlinie 2009/48/EG Sie stellen Spielzeug her, handeln mit diesen Produkten oder importieren sie? Wissen Sie Bescheid über die gesetzlichen Regelungen? Können Sie
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Arbeitskreis Europäische Normung und Qualitätssicherung
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Arbeitskreis Europäische Normung und Qualitätssicherung www.stmwi.bayern.de Aufgaben und Ziele des Arbeitskreises Europäische
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Arbeitskreis Europäische Normung und Qualitätssicherung www.stmwi.bayern.de Aufgaben und Ziele des Arbeitskreises Europäische
Merkblatt zur EU-Richtlinie. CE-Kennzeichnung. Kurzinformation EU-Richtlinien. Seite 1 von 9
 Merkblatt zur EU-Richtlinie 93/68/EWG Kurzinformation EU-Richtlinien Seite 1 von 9 Was müssen Hersteller über die wissen? Anfang der 90er Jahre sind die wesentlichen Voraussetzungen für die Vollendung
Merkblatt zur EU-Richtlinie 93/68/EWG Kurzinformation EU-Richtlinien Seite 1 von 9 Was müssen Hersteller über die wissen? Anfang der 90er Jahre sind die wesentlichen Voraussetzungen für die Vollendung
2. Abschnitt Begriffsbestimmungen
 Kurztitel Medizinproduktegesetz Kundmachungsorgan BGBl. Nr. 657/1996 zuletzt geändert durch BGBl. I Nr. 143/2009 Typ BG /Artikel/Anlage 2 Inkrafttretensdatum 21.03.2010 Abkürzung MPG Index 82/02 Gesundheitsrecht
Kurztitel Medizinproduktegesetz Kundmachungsorgan BGBl. Nr. 657/1996 zuletzt geändert durch BGBl. I Nr. 143/2009 Typ BG /Artikel/Anlage 2 Inkrafttretensdatum 21.03.2010 Abkürzung MPG Index 82/02 Gesundheitsrecht
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie. Anwendung von Normen im Rahmen der CE-Kennzeichnung
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Anwendung von Normen im Rahmen der CE-Kennzeichnung Die EU-Richtlinien, die die CE-Kennzeichnung vorschreiben, legen keine
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Anwendung von Normen im Rahmen der CE-Kennzeichnung Die EU-Richtlinien, die die CE-Kennzeichnung vorschreiben, legen keine
Anlage zur Akkreditierungsurkunde D-ZM nach DIN EN ISO/IEC :2015 i
 Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-12007-06-01 nach DIN EN ISO/IEC 17021-1:2015 i Gültigkeitsdauer: 07.02.2017 bis 14.07.2019 Ausstellungsdatum: 07.02.2017 Anlage
Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-12007-06-01 nach DIN EN ISO/IEC 17021-1:2015 i Gültigkeitsdauer: 07.02.2017 bis 14.07.2019 Ausstellungsdatum: 07.02.2017 Anlage
Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA Verordnung über Medizinprodukte Medizinprodukte-Verordnung
 juris Das Rechtsportal Gesamtes Gesetz Amtliche Abkürzung: MPV Ausfertigungsdatum: 20.12.2001 Gültig ab: 01.01.2002 Dokumenttyp: Rechtsverordnung Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA 7102-47-6
juris Das Rechtsportal Gesamtes Gesetz Amtliche Abkürzung: MPV Ausfertigungsdatum: 20.12.2001 Gültig ab: 01.01.2002 Dokumenttyp: Rechtsverordnung Quelle: Fundstelle: BGBl I 2001, 3854 FNA: FNA 7102-47-6
Merkblatt CE-Kennzeichen (französisch Communautés Européenes )
") Merkblatt CE-Kennzeichen (französisch Communautés Européenes ) Die CE-Kennzeichnung bestätigt die Konformität (Übereinstimmung) eines Erzeugnisses mit den in den europäischen Richtlinien nach der Neuen
Merkblatt CE-Kennzeichen (französisch Communautés Européenes ) Die CE-Kennzeichnung bestätigt die Konformität (Übereinstimmung) eines Erzeugnisses mit den in den europäischen Richtlinien nach der Neuen
Vorlesung Medizinrecht
 Vorlesung Medizinrecht Angewandte Gesundheitswissenschaften Teil 4: Arzneimittel- und Medizinprodukterecht Rechtsanwalt Wolfgang Wiefelspütz Fachanwalt für Medizinrecht Inhaltsangabe A. Einführung B. AMG
Vorlesung Medizinrecht Angewandte Gesundheitswissenschaften Teil 4: Arzneimittel- und Medizinprodukterecht Rechtsanwalt Wolfgang Wiefelspütz Fachanwalt für Medizinrecht Inhaltsangabe A. Einführung B. AMG
MEDICAL APPS. Software als Medizinprodukt
 MEDICAL APPS Software als Medizinprodukt MMag. Sabine Fehringer, LL.M. DLA Piper Weiss-Tessbach Rechtsanwälte GmbH 5. Mai 2017 0 Medical-Apps Gesundheits-Apps vermessen unsere Fitness analysieren physiologische
MEDICAL APPS Software als Medizinprodukt MMag. Sabine Fehringer, LL.M. DLA Piper Weiss-Tessbach Rechtsanwälte GmbH 5. Mai 2017 0 Medical-Apps Gesundheits-Apps vermessen unsere Fitness analysieren physiologische
Akkreditierungsumfang der Produktzertifizierungsstelle (EN ISO/IEC 17065:2012) TÜV AUSTRIA SERVICES GMBH / (Ident.Nr.: 0944)
 TÜV AUSTRIA SERVICES GMBH / (Ident.Nr.: 0944)") 1 2000/9/EG*2000/9/EC*2000/9/ 2000-03 Richtlinie 2000/9/EG des Europäischen Sicherheitsbauteile Anhang V CE Parlaments und des Rates vom 20. März 2000 über Seilbahnen für den Personenverkehr 2 2014/33/EU*2014/33/EU*2014/
1 2000/9/EG*2000/9/EC*2000/9/ 2000-03 Richtlinie 2000/9/EG des Europäischen Sicherheitsbauteile Anhang V CE Parlaments und des Rates vom 20. März 2000 über Seilbahnen für den Personenverkehr 2 2014/33/EU*2014/33/EU*2014/
Workshop Medizinprodukterecht. Peter Pieper Vistec AG
 Workshop Peter Pieper Vistec AG Was ist neu? Bisher: Richtlinie 93/42 EWG (Inverkehrbringen und Inbetriebnahme v. Med.-Produkten) Neu: MDR 2017 (Medical Device Regulation) (Europ. Vorschrift) Medizinproduktegesetz
Workshop Peter Pieper Vistec AG Was ist neu? Bisher: Richtlinie 93/42 EWG (Inverkehrbringen und Inbetriebnahme v. Med.-Produkten) Neu: MDR 2017 (Medical Device Regulation) (Europ. Vorschrift) Medizinproduktegesetz
RED Funkanlagen statt bisher R&TTE
 DVSI Information RED Funkanlagen statt bisher R&TTE (DVSI-Seminarthema am 10.04.15, 02.10.15, 07.04.16,001.12.16 und 06.04.17) Q u a l i t ä t s m a n a g e m e n t / Q u a l i t ä t s s i c h e r u n
DVSI Information RED Funkanlagen statt bisher R&TTE (DVSI-Seminarthema am 10.04.15, 02.10.15, 07.04.16,001.12.16 und 06.04.17) Q u a l i t ä t s m a n a g e m e n t / Q u a l i t ä t s s i c h e r u n
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie. Anwendung von Normen im Rahmen der CE-Kennzeichnung
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Anwendung von Normen im Rahmen der CE-Kennzeichnung Jene EU-Richtlinien, welche die CE-Kennzeichnung vorschreiben, legen
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Anwendung von Normen im Rahmen der CE-Kennzeichnung Jene EU-Richtlinien, welche die CE-Kennzeichnung vorschreiben, legen
Software als Medizinprodukt im Reformprozess
 Software als Medizinprodukt im Reformprozess 9. Augsburger Forum für Medizinprodukterecht Matthias Hölzer-Klüpfel Software für medizintechnische Geräte R&D Ausgaben 84 Mrd. 28 Mrd. Software 28 Mrd. 7 Mrd.
Software als Medizinprodukt im Reformprozess 9. Augsburger Forum für Medizinprodukterecht Matthias Hölzer-Klüpfel Software für medizintechnische Geräte R&D Ausgaben 84 Mrd. 28 Mrd. Software 28 Mrd. 7 Mrd.
Niederspannungsrichtlinie 2006/95/EG 1. GPSGV. Siemens AG Alle Rechte vorbehalten.
 Niederspannungsrichtlinie 2006/95/EG 1. GPSGV Hintergründe ganz allgemein Verbesserung / Beibehaltung des Sicherheitsniveaus Abbau von Handelshemmnissen. Gewährleistung des freien Warenverkehrs in der
Niederspannungsrichtlinie 2006/95/EG 1. GPSGV Hintergründe ganz allgemein Verbesserung / Beibehaltung des Sicherheitsniveaus Abbau von Handelshemmnissen. Gewährleistung des freien Warenverkehrs in der
Abgrenzung aus Sicht der Industrie - Medizinprodukte / BVMed e.v. -
 Abgrenzung aus Sicht der Industrie - Medizinprodukte / BVMed e.v. - BfArM im Dialog: Abgrenzung von Arzneimitteln Bonn, 19. September 2016 Dr. iur. Dr. med. Adem Koyuncu Rechtsanwalt und Arzt Assoz. Mitglied
Abgrenzung aus Sicht der Industrie - Medizinprodukte / BVMed e.v. - BfArM im Dialog: Abgrenzung von Arzneimitteln Bonn, 19. September 2016 Dr. iur. Dr. med. Adem Koyuncu Rechtsanwalt und Arzt Assoz. Mitglied
Medizinproduktegesetz (MPG)
") Einführung ins Medizinproduktegesetz (MPG) Prof. Dr. Christian Fegeler Zielsetzung und Rechtlicher Rahmen Medical Device Direktive (MDD) Richtlinie i 93/42/EWG sowie Richtlinien 90/385/EWG aktive implantierbare
Einführung ins Medizinproduktegesetz (MPG) Prof. Dr. Christian Fegeler Zielsetzung und Rechtlicher Rahmen Medical Device Direktive (MDD) Richtlinie i 93/42/EWG sowie Richtlinien 90/385/EWG aktive implantierbare
EG-Richtlinien und. Kälteanlagen. Bernhard Schrempf - KISC-KÄLTE-Information-Solution-Consulting
 EG-Richtlinien und Kälteanlagen 2012 www.kiscnet.com 1 Welche EG-Richtlinien sind in der Regel bei Kälteanlagen anzuwenden? www.kiscnet.com 2 2006/42/EG - Richtlinie über Maschinen und zur Änderung der
EG-Richtlinien und Kälteanlagen 2012 www.kiscnet.com 1 Welche EG-Richtlinien sind in der Regel bei Kälteanlagen anzuwenden? www.kiscnet.com 2 2006/42/EG - Richtlinie über Maschinen und zur Änderung der
Standalone Software. als Medizinprodukt. Matthias Hölzer-Klüpfel
 Standalone Software Standalone Software als Medizinprodukt Matthias Hölzer-Klüpfel Standalone Software MEDDEV 2.1/6 Klassifikation Beispiele Modules MDD 2007 Definition: Medizinprodukt [MDD, Artikel 1,
Standalone Software Standalone Software als Medizinprodukt Matthias Hölzer-Klüpfel Standalone Software MEDDEV 2.1/6 Klassifikation Beispiele Modules MDD 2007 Definition: Medizinprodukt [MDD, Artikel 1,
1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene Anwendung über einen Zeitraum von weniger als 60 Minuten bestimmt.
 Klassifizierung gemäß 93/42/EWG ANHANG IX KLASSIFIZIERUNGSKRITERIEN I. DEFINITONEN 1. Definitionen zu den Klassifizierungsregeln 1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene
Klassifizierung gemäß 93/42/EWG ANHANG IX KLASSIFIZIERUNGSKRITERIEN I. DEFINITONEN 1. Definitionen zu den Klassifizierungsregeln 1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene
0.2 Inhalt des Gesamtwerks 1
 1 Band 1 Vorwort Blick in die Zukunft Was ändert sich für Hersteller von Medizinprodukten? Die neue Verordnung (EU) Nr. 2017/745 über Medizinprodukte 1 Einführung 2 Anwendungsfristen und Übergangsbestimmungen
1 Band 1 Vorwort Blick in die Zukunft Was ändert sich für Hersteller von Medizinprodukten? Die neue Verordnung (EU) Nr. 2017/745 über Medizinprodukte 1 Einführung 2 Anwendungsfristen und Übergangsbestimmungen
Checkliste Technische Dokumentation Produktname
 Teil 1: Allgemeines Qualitätsmanagement Nr. Vorgelegte Dokumentation Anmerkung 1 Managementhandbuch 2 Spezielle Prozessanweisungen, Verfahrensanweisungen und Arbeitsanweisungen 3 Mitgeltende Formblätter
Teil 1: Allgemeines Qualitätsmanagement Nr. Vorgelegte Dokumentation Anmerkung 1 Managementhandbuch 2 Spezielle Prozessanweisungen, Verfahrensanweisungen und Arbeitsanweisungen 3 Mitgeltende Formblätter
Medizinprodukte-Sicherheitsplanverordnung
 Hygieneforum Sande 19.06.2014 Meldung von Vorkommnissen nach der Medizinproduke-Sicherheitsplanverordnung www.klinikum-bremen-ldw.de Klinikum Links der Weser Senator-Weßling-Str. 1 28277 Bremen Meldung
Hygieneforum Sande 19.06.2014 Meldung von Vorkommnissen nach der Medizinproduke-Sicherheitsplanverordnung www.klinikum-bremen-ldw.de Klinikum Links der Weser Senator-Weßling-Str. 1 28277 Bremen Meldung
Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen
 Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen build.well.being 2017 Andreas Böhler R n B Medical Software Consulting GmbH office@rnb-consulting.at build.well.being 2017 30.
Health Apps, Lifestyle Apps, Medical Apps Unterschiede und Rahmenbedingungen build.well.being 2017 Andreas Böhler R n B Medical Software Consulting GmbH office@rnb-consulting.at build.well.being 2017 30.
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen (PSA), Schutzvorrichtungen,
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 89/686/EWG Richtlinie über (PSA) Sie stellen persönliche Schutzausrüstungen (PSA), Schutzvorrichtungen,
Beschränkung der Verwendung von Gefahrstoffen in Elektro- und Elektronikgeräten (RoHS)
") Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Beschränkung der Verwendung von Gefahrstoffen in Elektro- und Elektronikgeräten (RoHS) Merkblatt zur EU-Richtlinie 2011/65/EU
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Beschränkung der Verwendung von Gefahrstoffen in Elektro- und Elektronikgeräten (RoHS) Merkblatt zur EU-Richtlinie 2011/65/EU
(Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION
 INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION") 15.7.2009 Amtsblatt der n Union C 163/1 IV (Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie
15.7.2009 Amtsblatt der n Union C 163/1 IV (Informationen) INFORMATIONEN DER ORGANE UND EINRICHTUNGEN DER EUROPÄISCHEN UNION KOMMISSION Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie
Richtlinie über Funkanlagen und Telekommunikationsendeinrichtungen
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Richtlinie über Funkanlagen und Telekommunikationsendeinrichtungen Merkblatt zur EU-Richtlinie 1999/5/EG Richtlinie über
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Richtlinie über Funkanlagen und Telekommunikationsendeinrichtungen Merkblatt zur EU-Richtlinie 1999/5/EG Richtlinie über
EU-Konformitätserklärungen
 EU-Konformitätserklärungen Eine EU-Konformitätserklärung sagt aus, dass das betreffende Gerät/das elektrische Betriebsmittel/die Funkanlage den Anforderungen der zutreffenden Richtlinien entspricht. Es
EU-Konformitätserklärungen Eine EU-Konformitätserklärung sagt aus, dass das betreffende Gerät/das elektrische Betriebsmittel/die Funkanlage den Anforderungen der zutreffenden Richtlinien entspricht. Es
Dipl.-Ing. Jürgen Bialek: zusätzliche Information zu Seminaren der Reihe integrated safety & compliance
 Dipl.-Ing. Jürgen Bialek: zusätzliche Information zu Seminaren der Reihe integrated safety & compliance Beispiele für EG-/EU-Konformitätserklärungen 1. EG-Konformitätserklärung nach Anhang II A der Richtlinie
Dipl.-Ing. Jürgen Bialek: zusätzliche Information zu Seminaren der Reihe integrated safety & compliance Beispiele für EG-/EU-Konformitätserklärungen 1. EG-Konformitätserklärung nach Anhang II A der Richtlinie
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie. Sicherheit von elektrischen Betriebsmitteln
 Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2014/35/EU Niederspannungsrichtlinie (Richtlinie über elektrische Betriebsmittel zur Verwendung
Bayerisches Staatsministerium für Wirtschaft und Medien, Energie und Technologie Merkblatt zur EU-Richtlinie 2014/35/EU Niederspannungsrichtlinie (Richtlinie über elektrische Betriebsmittel zur Verwendung
Merkblatt zur EU-Richtlinie 2009/48/EG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Richtlinie über die Sicherheit von Spielzeug Merkblatt zur EU-Richtlinie 2009/48/EG Sicherheit von Spielzeug Sie stellen
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Richtlinie über die Sicherheit von Spielzeug Merkblatt zur EU-Richtlinie 2009/48/EG Sicherheit von Spielzeug Sie stellen
Gesetzliche Vorgaben für die Aufbereitung von Instrumenten in der Podologie
 Gesetzliche Vorgaben für die Aufbereitung von Instrumenten in der Podologie Rechtliche Abgrenzung zwischen Podologie und Fußpflege Ziele des Medizinprodukterechtes: für die Gesundheit und den erforderlichen
Gesetzliche Vorgaben für die Aufbereitung von Instrumenten in der Podologie Rechtliche Abgrenzung zwischen Podologie und Fußpflege Ziele des Medizinprodukterechtes: für die Gesundheit und den erforderlichen
Anlage zur Akkreditierungsurkunde D-ZM nach DIN EN ISO/IEC :2015
 Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-17591-01-00 nach DIN EN ISO/IEC 17021-1:2015 Gültigkeitsdauer: 14.08.2017 bis 26.05.2020 Ausstellungsdatum: 14.08.2017 Urkundeninhaber:
Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-17591-01-00 nach DIN EN ISO/IEC 17021-1:2015 Gültigkeitsdauer: 14.08.2017 bis 26.05.2020 Ausstellungsdatum: 14.08.2017 Urkundeninhaber:
Konformitätsbewertung 3.9 A 3
 Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 3 Reihenfolge bei der Durchführung von Konformitätsbewertungsverfahren Artikel 11 der Richtlinie 93/42/EWG legt fest, welche Konformitätsbewertungsverfahren
Antworten und Beschlüsse des EK-Med Konformitätsbewertung 3.9 A 3 Reihenfolge bei der Durchführung von Konformitätsbewertungsverfahren Artikel 11 der Richtlinie 93/42/EWG legt fest, welche Konformitätsbewertungsverfahren
Die Kombination von Medizinprodukten. SystemCheck
 Die Kombination von Medizinprodukten SystemCheck Fachtagung der FKT 12.06.2008 Untertitel Die Prüfung und Bewertung von medizinischen elektrischen Systemen mit rechtssicherer Dokumentation zum Schutz von
Die Kombination von Medizinprodukten SystemCheck Fachtagung der FKT 12.06.2008 Untertitel Die Prüfung und Bewertung von medizinischen elektrischen Systemen mit rechtssicherer Dokumentation zum Schutz von
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie. Medizinprodukte. Merkblatt zur EU-Richtlinie 93/42/EWG
 Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 93/42/EWG Stand: März 2005 Richtlinie über Sie stellen oder Zubehör her, handeln mit diesen
Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie Merkblatt zur EU-Richtlinie 93/42/EWG Stand: März 2005 Richtlinie über Sie stellen oder Zubehör her, handeln mit diesen
Know-How für die Medizintechnik
 CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
Gesamte Rechtsvorschrift für Konformitätsbewertung von Medizinprodukten, Fassung vom
 Gesamte Rechtsvorschrift für Konformitätsbewertung von Medizinprodukten, Fassung vom 07.03.2014 Langtitel Verordnung der Bundesministerin für Gesundheit und Frauen über die Konformitätsbewertung von Medizinprodukten
Gesamte Rechtsvorschrift für Konformitätsbewertung von Medizinprodukten, Fassung vom 07.03.2014 Langtitel Verordnung der Bundesministerin für Gesundheit und Frauen über die Konformitätsbewertung von Medizinprodukten
CE-Kennzeichnung bei Medizinprodukten und Hilfsmitteln
 CE-Kennzeichnung bei Medizinprodukten und Hilfsmitteln ÜBERSICHT 1. Einführung 2. CE-Kennzeichnung bei Medizinprodukten 3. CE-Kennzeichnung bei Hilfsmitteln 4. Hilfsmittelverzeichnis und CE-Kennzeichnung
CE-Kennzeichnung bei Medizinprodukten und Hilfsmitteln ÜBERSICHT 1. Einführung 2. CE-Kennzeichnung bei Medizinprodukten 3. CE-Kennzeichnung bei Hilfsmitteln 4. Hilfsmittelverzeichnis und CE-Kennzeichnung
Beschaffung von Medizinprodukten
 Beschaffung von Medizinprodukten Dipl.-Ing. Franz Josef Fegerl TÜV Österreich, Leiter des Institutes für Medizintechnik Das österreichische Medizinproduktegesetz (MPG) BGBl.-Nr. 657/1996, geändert durch
Beschaffung von Medizinprodukten Dipl.-Ing. Franz Josef Fegerl TÜV Österreich, Leiter des Institutes für Medizintechnik Das österreichische Medizinproduktegesetz (MPG) BGBl.-Nr. 657/1996, geändert durch
Med-Info. Richtlinie 93/42/EWG über Medizinprodukte. TÜV SÜD Product Service GmbH. Internationale Fach-Informationen für die Medizinproduktebranche
 Med-Info Internationale Fach-Informationen für die Medizinproduktebranche Richtlinie 93/42/EWG über Medizinprodukte Praxisorientierte Zusammenfassung der wichtigsten Aspekte und Anforderungen der Richtlinie
Med-Info Internationale Fach-Informationen für die Medizinproduktebranche Richtlinie 93/42/EWG über Medizinprodukte Praxisorientierte Zusammenfassung der wichtigsten Aspekte und Anforderungen der Richtlinie
Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG. Tamara Bauer
 Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG Tamara Bauer Inhalt Definitionen Richtlinien Kombinationsprodukte 2 Fallbeispiele: Medizinprodukt/Arzneimittel Medizinprodukt/Arzneimittel
Kombinationsprodukte im Kontext der Revision der RL 93/42/EWG Tamara Bauer Inhalt Definitionen Richtlinien Kombinationsprodukte 2 Fallbeispiele: Medizinprodukt/Arzneimittel Medizinprodukt/Arzneimittel
MEDIZINPRODUKTEABGABENVERORDNUNG
 MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
GV 9 Achte Verordnung zum Geräte- Produktsicherheitsgesetz (Verordnung über das Inverkehrbringen von persönlichen Schutzausrüstungen - 8.
 GV 9 Achte Verordnung zum Geräte- Produktsicherheitsgesetz (Verordnung über das Inverkehrbringen von persönlichen Schutzausrüstungen - 8. GPSGV) in der Fassung der Bekanntmachung vom 20. Februar 1997 (BGBl.
GV 9 Achte Verordnung zum Geräte- Produktsicherheitsgesetz (Verordnung über das Inverkehrbringen von persönlichen Schutzausrüstungen - 8. GPSGV) in der Fassung der Bekanntmachung vom 20. Februar 1997 (BGBl.
CE-ZERTIFIZIERUNG VON MASCHINEN FASI-VORTRAGSVERANSTALTUNG - BRANDSCHUTZ UND MASCHINENSICHERHEIT
 FASI-VORTRAGSVERANSTALTUNG - BRANDSCHUTZ UND MASCHINENSICHERHEIT Andreas Siegmund (BGV) Referat Produktsicherheit - V22-21.03.2017 Marktüberwachung in Hamburg durch das Referat Produktsicherheit Aufgabengebiet
FASI-VORTRAGSVERANSTALTUNG - BRANDSCHUTZ UND MASCHINENSICHERHEIT Andreas Siegmund (BGV) Referat Produktsicherheit - V22-21.03.2017 Marktüberwachung in Hamburg durch das Referat Produktsicherheit Aufgabengebiet
CE-Kennzeichnung Grundlagen - Pflichten
 CE-Kennzeichnung Grundlagen - Pflichten 1 Inhalte 1 2 3 Pflichten der Wirtschaftsakteure im Zusammenhang mit der CE-Kennzeichnung CE-Richtlinien Schritte zur CE-Kennzeichnung 2 1 Inhalte 1 2 3 Pflichten
CE-Kennzeichnung Grundlagen - Pflichten 1 Inhalte 1 2 3 Pflichten der Wirtschaftsakteure im Zusammenhang mit der CE-Kennzeichnung CE-Richtlinien Schritte zur CE-Kennzeichnung 2 1 Inhalte 1 2 3 Pflichten
GV09 Gesetze und Verordnungen
 Achte Verordnung zum Produktsicherheitsgesetz (Verordnung über die Bereitstellung von persönlichen Schutzausrüstungen auf dem Markt - 8. ProdSV) in der Fassung der Bekanntmachung vom 20. Februar 1997 (BGBl.
Achte Verordnung zum Produktsicherheitsgesetz (Verordnung über die Bereitstellung von persönlichen Schutzausrüstungen auf dem Markt - 8. ProdSV) in der Fassung der Bekanntmachung vom 20. Februar 1997 (BGBl.
Amtsblatt L390 der Europäischen Union vom 31. Dezember
 Fachinformation Nr.: 2013-08 MeßTechnikNord GmbH Team Jena Prüssingstraße 41 07745 Jena Telefon: 03641-65-3780 Fax: 03641-65-3927 E-Mail: info@messtechniknord.de Internet http://www.messtechniknord.de
Fachinformation Nr.: 2013-08 MeßTechnikNord GmbH Team Jena Prüssingstraße 41 07745 Jena Telefon: 03641-65-3780 Fax: 03641-65-3927 E-Mail: info@messtechniknord.de Internet http://www.messtechniknord.de
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH. Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II
 1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
Medizinprodukte sicher betreiben und anwenden Regionaler Arbeitskreis für Arbeitssicherheit Oldenburg
 www.bgw-online.de Medizinprodukte sicher betreiben und anwenden BGW Präventionsdienste Delmenhorst Roter Faden Warum Medizinprodukte Anforderungen an Medizinprodukte Anwender- und Betreiberpflichten Überblick
www.bgw-online.de Medizinprodukte sicher betreiben und anwenden BGW Präventionsdienste Delmenhorst Roter Faden Warum Medizinprodukte Anforderungen an Medizinprodukte Anwender- und Betreiberpflichten Überblick
Besonderheiten bei der Abgrenzung von Medizinprodukten
 Besonderheiten bei der Abgrenzung von Medizinprodukten Dr. Nicole Rämsch-Günther Dr. N. Rämsch-Günther BfArM im Dialog: Abgrenzung von Arzneimitteln Besonderheiten bei der Abgrenzung von Medizinprodukten
Besonderheiten bei der Abgrenzung von Medizinprodukten Dr. Nicole Rämsch-Günther Dr. N. Rämsch-Günther BfArM im Dialog: Abgrenzung von Arzneimitteln Besonderheiten bei der Abgrenzung von Medizinprodukten
Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen
 Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen Risiken erkennen, minimieren, finanzieren heute und morgen SAQ Fachgruppe Medizinprodukte Hersteller, 22.3.2007, Zofingen Monika Gattiker
Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen Risiken erkennen, minimieren, finanzieren heute und morgen SAQ Fachgruppe Medizinprodukte Hersteller, 22.3.2007, Zofingen Monika Gattiker
Praktische Umsetzung in der Handhabung von Medizinprodukten
 Praktische Umsetzung in der Handhabung von Medizinprodukten Andreas Clarin Hygieneakademie Ruhr, Essen Definition Medizinprodukte ( 3 MPG) Medizinprodukte sind alle Instrumente, Apparate, Vorrichtungen,
Praktische Umsetzung in der Handhabung von Medizinprodukten Andreas Clarin Hygieneakademie Ruhr, Essen Definition Medizinprodukte ( 3 MPG) Medizinprodukte sind alle Instrumente, Apparate, Vorrichtungen,
Beschaffung von Medizinprodukten DIVI Jochen Kaiser Klinikum Stuttgart
 Beschaffung von Medizinprodukten DIVI 2015 Jochen Kaiser Klinikum Stuttgart J.Kaiser@klinikum-stuttgart.de Jochen.kaiser@medsias.de Was ist das? Problem: asynchrone Beschaffungsmechanismen Problem: asynchrone
Beschaffung von Medizinprodukten DIVI 2015 Jochen Kaiser Klinikum Stuttgart J.Kaiser@klinikum-stuttgart.de Jochen.kaiser@medsias.de Was ist das? Problem: asynchrone Beschaffungsmechanismen Problem: asynchrone
Originaltext: 8. GSGV
 Originaltext: 8. GSGV Daten des Gesetzes Einleitung Auf den folgenden Seiten finden Sie den Originaltext der 8. Verordnung zum Gerätessicherheitsgesetz. Titel Verordnung über das Inverkehrbringen von persönlichen
Originaltext: 8. GSGV Daten des Gesetzes Einleitung Auf den folgenden Seiten finden Sie den Originaltext der 8. Verordnung zum Gerätessicherheitsgesetz. Titel Verordnung über das Inverkehrbringen von persönlichen
ANHANG VORSCHLAG FÜR EINE VERORDNUNG DES EUROPÄISCHEN PARLAMENTS UND DES RATES
 EUROPÄISCHE KOMMISSION Brüssel, den 19.12.2017 COM(2017) 795 final ANNEX 1 ANHANG VORSCHLAG FÜR EINE VERORDNUNG DES EUROPÄISCHEN PARLAMENTS UND DES RATES zur Festlegung von Bestimmungen und Verfahren für
EUROPÄISCHE KOMMISSION Brüssel, den 19.12.2017 COM(2017) 795 final ANNEX 1 ANHANG VORSCHLAG FÜR EINE VERORDNUNG DES EUROPÄISCHEN PARLAMENTS UND DES RATES zur Festlegung von Bestimmungen und Verfahren für