Risikomanagement nach EN ISO 14971:2007
|
|
|
- Tobias Schulz
- vor 8 Jahren
- Abrufe
Transkript
1 , FMEA, Risikoanalysen, QM, SQ, SCIO Risikomanagement nach EN ISO 14971:2007 DGQ Regionalkreis Bodensee-Oberschwaben am Mader 1, Waldburg +49(0)7529/
7529/634567 www.")
2 Inhalt Grundlagen Risikomanagementprozess Dokumente Organisation (QM-System, operative Prozesse)

3 Grundlagen Definition Medizinprodukt Einzeln oder miteinander verbundene Instrumente, Apparate, Vorrichtungen, Stoffe oder andere Gegenstände einschließlich der für ein einwandfreies Funktionieren des Medizinproduktes eingesetzten Software, die vom Hersteller zur Anwendung für Menschen für folgende Zwecke bestimmt sind: Erkennung, Verhütung, Erfassung, Behandlung oder Linderung von Krankheiten Erkennung, Erfassung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen Untersuchung, Ersatz oder Veränderung des anatomischen Aufbaus oder eines physiologischen Vorgangs Lebenserhaltung und Lebensunterstützung Desinfektion von Medizinprodukten Bereitstellung von Informationen für medizinische Zwecke mittels In-vitro-Untersuchung von Probestücken des menschlichen Körpers Empfängnisregelung

4 Grundlagen Grundlegende Anforderungen der EU-Richtlinien und Normen Richtlinie 93/42/EG Medizinprodukte müssen für Patienten, Anwender und Dritte einen hochgradigen Schutz bieten und die vom Hersteller angegebenen Leistungen erfüllen. Richtlinie 2007/47/EG Da die Hersteller die Entwicklung und die Herstellung von Medizinprodukten immer häufiger bei Dritten in Auftrag geben, muss der Hersteller unbedingt nachweisen, dass er jene Dritten angemessenen Kontrollen unterzieht, um dauerhaft zu gewährleisten, dass das Qualitätssicherungssystem effizient arbeitet. EN ISO (7.1 Produktrealisierung) Die Organisation muss dokumentierte Anforderungen für das Risikomanagement während der gesamten Produktrealisierung erarbeiten. Es müssen Aufzeichnungen geführt werden, die sich aus dem Risikomanagement ergeben. EN (2007) *RISIKOMANAGEMENT-PROZESS bei ME-GERÄTEN oder ME-SYSTEMEN Es muss ein RISIKOMANAGEMENT-PROZESS nach ISO durchgeführt werden.

5 Anforderungen 2007/47/EG Für Produkte der Klasse I muss der Hersteller die Anforderungen des Anhang I und VII einhalten Anhang I: Die Produkte müssen so ausgelegt und hergestellt sein, dass die Sicherheit von Patienten, Anwender oder Dritte nicht gefährdet ist Weitgehendste Verringerung von Anwendungsfehler Berücksichtigung der Kenntnisse des Anwenders Integration des Sicherheitskonzepts in die Entwicklung und den Bau des Produkts Es sind vom Hersteller folgende Grundsätze zu berücksichtigen: Beseitigung oder Minimierung von Risiken Ggf. ergreifen von angemessenen Schutzmaßnahmen Unterrichtung der Benutzer über die Restrisiken

6 Anforderungen 2007/47/EG Anhang VII Die technische Dokumentation muss die Bewertung der Konformität des Produktes mit den Anforderungen der Richtlinie ermöglichen Produktdokumentation (Zeichnungen, Beschreibungen, ) Risikoanalyse Liste der Normen Ergebnisse von Konstruktionsberechnungen und Prüfungen Kennzeichnung und Gebrauchsanweisung.

7 Risikomanagementprozess Risikoanalyse Risikobewertung Risikobeherrschung (Risiko-Nutzen- Analyse) Bewertung der Akzeptanz des Gesamt- Restrisikos Risikomanagementbericht Informationen aus der Herstellung und der Herstellung nachgelagerten Phasen.

8 Risikomanagementprozess Risikomanagementprozess umfasst den gesamten Lebenszyklus des Produktes und muss folgendes enthalten Design Herstellung (incl. Sterilisation, Verpackung, Kennzeichnung, Lagerung, Handhabung/Transport und Vertrieb) Der Herstellung nachgelagerter Phasen (Anwendung)

9 Risikoanalyse

10 Risikoanalyse Bei der Funktion-/Anforderungsanalyse sind z.b. zu berücksichtigen: Anwendung des Produktes Tätigkeiten mit dem Produkt Chemische, physikalische und biologische Eigenschaften Inhalte von Gebrauchsanweisungen Kennzeichnung und Verpackung der Produkte Zu beachten sind: Verträglichkeit der Werkstoffe Energieeintrag-/entzug Sterilität; Reinigung Umgebungsbedingungen Messgenauigkeit Umweltverträglichkeit Wartung, Kalibrierung Nutzungsdauer, Zuverlässigkeit Bedienbarkeit Informationen.

11 Risikoanalyse

12 Risikoanalyse Gefährdungsanalyse Zusammenstellung der bekannten und vorhersehbaren Gefährdungen sowohl unter Normal- wie unter Fehlerbedingungen Nichteinhaltung der geforderten Eigenschaften Berücksichtung von einschlägigen Normen (z.b. Gefährdungen gemäß EN )

13 Risikoanalyse Beispiel für Gefährdungen Beispiele energetischer Gefährd. Beispiele biol. oder chem. Gefährd. Beispiele v. Gefährd. durch den Betrieb Beispiele v. Gefährd. durch Informationen IR-Strahlung Kontamination Falschmessungen Falsche Spezifikation Temperatur Reizwirkung Falsche Aussagen/ Anzeigen Falsche / unzureichende Anweisungen Elektrische Felder Bakterien Fehlbedienung Fehlende Warnhinweise

14 Risikoanalyse
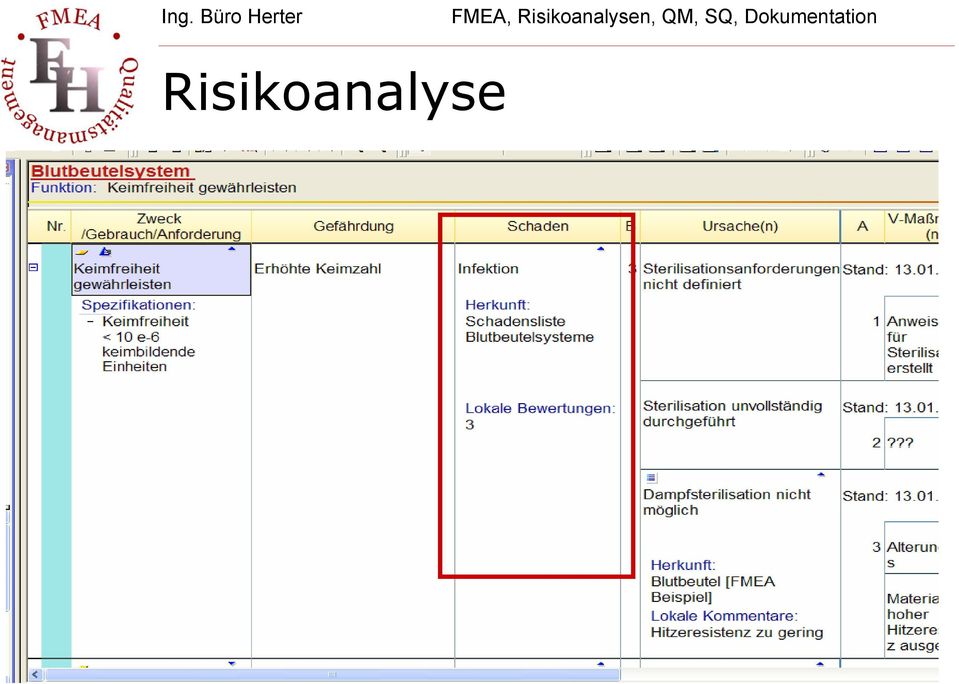
15 Risikoanalyse Schadensanalyse Ist eine Wahrscheinlichkeit vorhanden, dass eine Gefährdung auftreten kann (Gefährdungssituation) so kann daraus ein Schaden entstehen. Es sind wiederum Schäden an: Patienten Anwender Dritte zu berücksichtigen

16 Risikoanalyse

17 Risikoanalyse Ursachenanalyse Welche Fehler können zu einer Gefährdung bzw. zu einer Gefährdungssituation führen? Design- / Auslegungsfehler (z.b falsch bemessenes Bauteil) Herstellprozessfehler (z.b Verwendung unzulässiger Hilfsstoffe) Anwendungsfehler (z.b. Ausbildung entspricht nicht Anwendungsvoraussetzung) Servicefehler (z.b. falsche Sicherung eingesetzt) Dokumentationsfehler
 Anwendungsfehler (z.b. Ausbildung entspricht nicht Anwendungsvoraussetzung) Servicefehler (z.")
18 Risikoanalyse
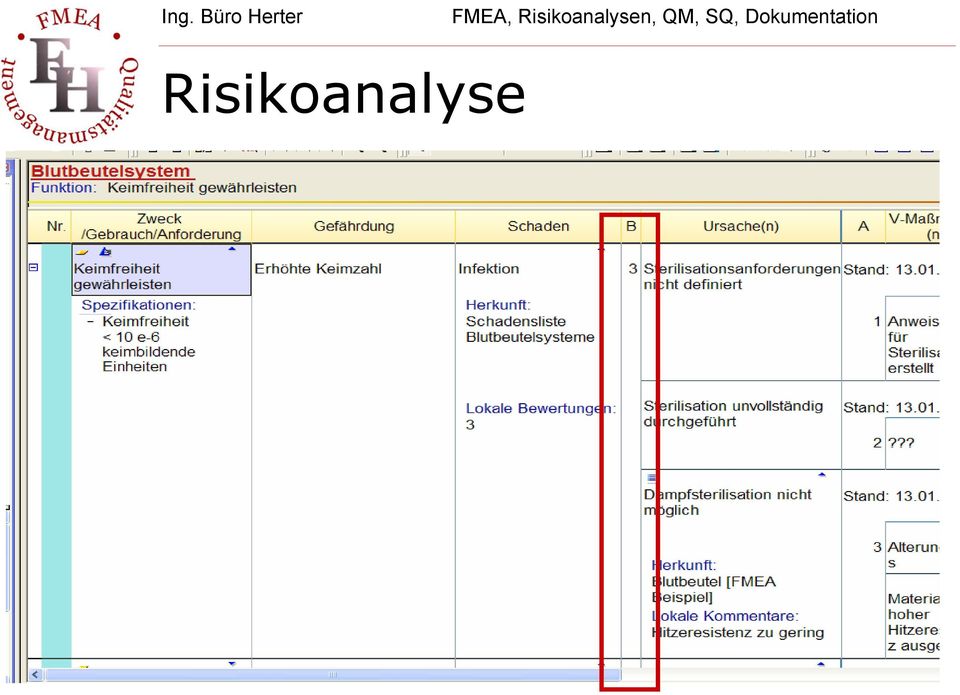
19 Risikoanalyse Bsp. einer Schweregradeinstufung des Schadens katastrophal Führt zum Tode des Patienten kritisch ernst gering vernachlässigbar Führt zu dauernder Behinderung oder einer lebensbedrohlichen Schädigung Führt zu einer Schädigung oder Behinderung, die ein sachkundiges medizinisches Eingreifen erfordert Führt zu einer zeitweiligen Schädigung oder Behinderung, die kein sachkundiges medizinisches Eingreifen erfordert Unannehmlichkeiten oder zeitweilige Beschwerden

20 Risikoanalyse

21 Risikoanalyse P1 ist die Wahrscheinlichkeit des Auftretens einer Gefährdungssituation. P2 ist die Wahrscheinlichkeit einer Gefährdungssituation, die zum Schaden führt. B -Bewertung A -Bewertung A X B
22 Risikoanalyse Einschätzung des Risikos Auftretenswahscheinlichkeit Die Einschätzung der Wahrscheinlichkeit bezieht die Umstände vom Auftreten der Ursache bis zum Auftreten des Schadens ein. Es sind folgende Fragen zu beantworten: Tritt die Gefährdungssituation ohne Vorliegen eines Versagens auf? Tritt die Gefährdungssituation bei einem Versagensmodus auf? Tritt die Gefährdungssituation nur unter Bedingungen mehrfachen Versagens auf? Wie wahrscheinlich ist es, dass eine Gefährdungssituation zu einem Schaden führt?
23 Risikobewertung
24 Risikobeherrschung Wird ein Risiko als inakzeptabel eingestuft, so müssen geeignete Maßnahmen zur Risikovermeidung oder Minimierung umgesetzt werden. Maßnahmen zur Erhöhung der Sicherheit können sein: direkte Sicherheit durch das Design (z.b. Designänderung) Schutzmaßnahmen im Medizinprodukt selbst (z.b. Alarme) Sicherheit durch den Herstellungsprozess (z.b. Prozessabsicherung) Informationen zur Sicherheit (z.b. Betriebsanleitung)
25 Risikobeherrschung Es ist zu prüfen ob Maßnahmen umgesetzt wurden Die umgesetzte Maßnahme das Risiko verringert Durch die Maßnahme keine weiteres Risiko entsteht, bzw. das Risiko anderer Merkmale / Gefährdungen erhöht Können Risiken nicht vollständig eliminiert werden, so ist eine Risiko-Nutzen-Analyse durchzuführen Überwiegt der Nutzen des Medizinproduktes das Risiko, muss der Hersteller entscheiden, welche Informationen zur Sicherheit notwendig sind. Grundlage: Daten Literatur
26 Risikobeherrschung Risiken durch Maßnahmen Einführung neuer Risiken durch die Maßnahmen zur Reduzierung anderer Risiken Konstruktive Änderungen Prozessänderungen Überprüfung inwiefern Maßnahmen Funktionen, den bestimmungsgemäßen Gebrauch oder die Merkmale des Produktes verändern
27 Risikomanagementbericht Der Risikomanagementbericht muss Nachweise zu folgenden Punkten beinhalten: Risikomanagementplan wurde implementiert Gesamt-Restrisiko ist akzeptabel Methoden um relevante Informationen aus der Herstellung und Herstellung nachgelagerten Phasen zu erhalten
28
29 Risikomanagementplan Beschreibung des Medizinproduktes Phasen des Lebenszyklus des Medizinproduktes Projektphasen / relevante Phasen für das Risikomanagement Verantwortliche und Befugnisse Teammitglieder, Freigabeberechtigte Anforderungen an die Überprüfung des Risikomanagements Kriterien, Zeitpunkt von Reviews Kriterien für die Akzeptanz der Risiken Risikomatrix Tätigkeiten der Verifizierung Wann werden welche Prüfungen durchgeführt Information aus der Herstellung nachgelagerten Phasen Verweis auf VA, Prozesse und ggf. Produktspezifische Anforderungen => Verweise auf vorhandene Dokumente reicht oft aus
30 Risikomanagementakte Inhalt: Allgemeiner Teil Risikomanagementplan Risikomanagementprozess Verantwortung der Leitung Anwendbare Gesetze, Vorschriften, Richtlinien, Normen, Risikomanagementbericht die Risikoanalyse (Gebrauch/Zweck, Gefährdungen) die Risikobewertung; die Implementierung und Verifizierung der Maßnahmen zur Risikobeherrschung; die Beurteilung der Akzeptanz jedes Restrisikos Nachgelagerte Phasen Interne / externe Reklamationen Produkt- / Prozessänderungen Marktbeobachtung Die Risikomanagementakte kann auf vorhandene Dokumente verweisen
31 Verantwortung der Leitung Nachweis der Verpflichtung zum Risikomanagementprozess Verfügbarkeit geeigneter Ressourcen Beauftragung von qualifiziertem Personal Politik zur Festlegung der Kriterien für die Akzeptanz von Risiken Überprüfung der Eignung des Risikomanagementprozesses Dokumentation von Maßnahmen hinsichtlich des Risikomanagements Umsetzung im QM-System durch z.b. Erstellung einer Risikopolitik Integration der Themen in das QM-Handbuch Kapitel Verantwortung der Leitung Aufnahme des Themas in das Management-Review
32 Qualifikation des Personals Angemessenes Wissen des Personals bezüglich: Risikomanagement Produkt Produktanwendung Aufzeichnungen der Schulungen
33 Nachgelagerte Phasen Nachgelagerte Phasen beinhalten Kundenreklamationen Feldbeobachtungsergebnisse (Anwender, Service, ) Designänderungen/-optimierungen Prozessänderungen/-optimierungen Änderung der Anforderungen (Normen, ) Umsetzung im QM-System durch: Änderungsmanagementprozess (Produkt, Prozess) Reklamationsablauf Einführung / Änderung von Normen, Gesetzen usw. Überprüfung der Sicherheitsrelevanz Aktualisierung der Risikoanalyse Aktualisierung der Risikomanagementakte
34 Für Ihre Aufmerksamkeit bedanke ich mich und stehe Ihnen für Fragen gerne Zur Verfügung Ing. Büro Herter Mader 1, Waldburg, Tel.:
35 Links, Dokumente 2007_47_EG 93_42_EWG Leitfaden zur Medizinprodukte-Regulierung Medizinproduktegesetz MPG
Risikomanagement bei Medizinprodukten
 Risikomanagement bei Medizinprodukten 10. Jahrestagung der AAL 24./25. September 2010 Stuttgart 2010 mdc medical device certification GmbH Risikomanagement 1 Regulatorische Grundlagen Richtlinie 93/42/EWG
Risikomanagement bei Medizinprodukten 10. Jahrestagung der AAL 24./25. September 2010 Stuttgart 2010 mdc medical device certification GmbH Risikomanagement 1 Regulatorische Grundlagen Richtlinie 93/42/EWG
3 Berücksichtigung anerkannter Standards / Normen / Nachweise 3.1 Methoden des Risikomanagements
 Datum: Unternehmen: Adresse: Straße, PLZ, Ort Produkt Auditor/in: Name Unterschrift Bezeichnung 1 Anwendungsbereich xxxxxxxx Diese Checkliste dient dem Auditteam als Vorgabe und Hilfe zur sachgerechten
Datum: Unternehmen: Adresse: Straße, PLZ, Ort Produkt Auditor/in: Name Unterschrift Bezeichnung 1 Anwendungsbereich xxxxxxxx Diese Checkliste dient dem Auditteam als Vorgabe und Hilfe zur sachgerechten
Medizintechnik und Informationstechnologie im Krankenhaus. Dr. Andreas Zimolong
 Medizintechnik und Informationstechnologie im Krankenhaus Dr. Andreas Zimolong DIN EN 80001-1:2011 Anwendung des Risikomanagements für IT-Netzwerke, die Medizinprodukte beinhalten Teil 1: Aufgaben, Verantwortlichkeiten
Medizintechnik und Informationstechnologie im Krankenhaus Dr. Andreas Zimolong DIN EN 80001-1:2011 Anwendung des Risikomanagements für IT-Netzwerke, die Medizinprodukte beinhalten Teil 1: Aufgaben, Verantwortlichkeiten
Software. Martin Zauner. martin.zauner@fh-linz.at +43 (0)732 804 52100. FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz
732 804 52100. FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz") Martin Zauner martin.zauner@fh-linz.at +43 (0)732 804 52100 FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz 1 Gesundheitswesen Zusammenwachsen zweier Welten bringt neue Herausforderungen
Martin Zauner martin.zauner@fh-linz.at +43 (0)732 804 52100 FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz 1 Gesundheitswesen Zusammenwachsen zweier Welten bringt neue Herausforderungen
Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de
 ISO/IEC 62304 Medizingeräte-Software Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de DQS Medizin nprodukte GmbH Übersicht Basics Wann ist ein MP Software? Markteinführung vor der 62304 alles
ISO/IEC 62304 Medizingeräte-Software Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de DQS Medizin nprodukte GmbH Übersicht Basics Wann ist ein MP Software? Markteinführung vor der 62304 alles
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten
 Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") 05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
EN 80001-1. Risikomanagement für IT-Netzwerke, die Medizinprodukte beinhalten
 EN 80001-1 Risikomanagement für IT-Netzwerke, die Medizinprodukte beinhalten Kontakt: gsm Gesellschaft für Sicherheit in der Medizintechnik GmbH Ing. Lukas Dolesch Leitermayergasse 43 1180 Wien Tel.: 0043
EN 80001-1 Risikomanagement für IT-Netzwerke, die Medizinprodukte beinhalten Kontakt: gsm Gesellschaft für Sicherheit in der Medizintechnik GmbH Ing. Lukas Dolesch Leitermayergasse 43 1180 Wien Tel.: 0043
Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten
 Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten Alexander Fink Hochschule Mannheim SS 2013 A. Fink Seite 1 Steigen wir ein Wo und wie kommt der Medizintechnik-Ingenieur
Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten Alexander Fink Hochschule Mannheim SS 2013 A. Fink Seite 1 Steigen wir ein Wo und wie kommt der Medizintechnik-Ingenieur
Abschnitt 1 Anwendungsbereich und Allgemeine Anforderungen an die Konformitätsbewertung 1 Anwendungsbereich
 13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt?
 DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
Software-Entwicklungsprozesse zertifizieren
 VDE-MedTech Tutorial Software-Entwicklungsprozesse zertifizieren Dipl.-Ing. Michael Bothe, MBA VDE Prüf- und Zertifizierungsinstitut GmbH BMT 2013 im Grazer Kongress 19.09.2013, 10:00-10:30 Uhr, Konferenzraum
VDE-MedTech Tutorial Software-Entwicklungsprozesse zertifizieren Dipl.-Ing. Michael Bothe, MBA VDE Prüf- und Zertifizierungsinstitut GmbH BMT 2013 im Grazer Kongress 19.09.2013, 10:00-10:30 Uhr, Konferenzraum
Managementbewertung Managementbewertung
 Managementbewertung Grundlagen für die Erarbeitung eines Verfahrens nach DIN EN ISO 9001:2000 Inhalte des Workshops 1. Die Anforderungen der ISO 9001:2000 und ihre Interpretation 2. Die Umsetzung der Normanforderungen
Managementbewertung Grundlagen für die Erarbeitung eines Verfahrens nach DIN EN ISO 9001:2000 Inhalte des Workshops 1. Die Anforderungen der ISO 9001:2000 und ihre Interpretation 2. Die Umsetzung der Normanforderungen
Entwurf. Anwendungsbeginn E DIN EN 62304 (VDE 0750-101):2013-10. Anwendungsbeginn dieser Norm ist...
:2013-10. Anwendungsbeginn dieser Norm ist...") Anwendungsbeginn Anwendungsbeginn dieser Norm ist.... Inhalt Einführung... 13 1 Anwendungsbereich... 16 1.1 *Zweck... 16 1.2 *Anwendungsbereich... 16 1.3 Beziehung zu anderen Normen... 16 1.4 Einhaltung...
Anwendungsbeginn Anwendungsbeginn dieser Norm ist.... Inhalt Einführung... 13 1 Anwendungsbereich... 16 1.1 *Zweck... 16 1.2 *Anwendungsbereich... 16 1.3 Beziehung zu anderen Normen... 16 1.4 Einhaltung...
Der Schutz von Patientendaten
 Der Schutz von Patientendaten beim Einsatz von Medizinprodukten aus Betreibersicht 17.06.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
Der Schutz von Patientendaten beim Einsatz von Medizinprodukten aus Betreibersicht 17.06.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
Software als Medizinprodukt
 Software als Medizinprodukt DI Dr. Gerhard Wrodnigg, MSc. TÜV AUSTRIA SERVICES Software als Medizinprodukt Wann ist Software ein Medizinprodukt? Änderung der RL 93/42/EWG durch 2007/47/EG Qualification
Software als Medizinprodukt DI Dr. Gerhard Wrodnigg, MSc. TÜV AUSTRIA SERVICES Software als Medizinprodukt Wann ist Software ein Medizinprodukt? Änderung der RL 93/42/EWG durch 2007/47/EG Qualification
Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar?
 Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar? 04.06.2013 Medical Apps 2013 Kathrin Schürmann, Rechtsanwältin 1 2013 ISiCO Datenschutz GmbH All rights reserved 2 1 Chancen und Risiken
Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar? 04.06.2013 Medical Apps 2013 Kathrin Schürmann, Rechtsanwältin 1 2013 ISiCO Datenschutz GmbH All rights reserved 2 1 Chancen und Risiken
Qualitätsmanagement- Handbuch nach DIN EN ISO 13485:2010-01 prozessorientiert
 Qualitätsmanagement- nach DIN EN ISO 13485:2010-01 prozessorientiert Version 0 / Exemplar Nr.: QMH unterliegt dem Änderungsdienst: x Informationsexemplar: Hiermit wird das vorliegende für gültig und verbindlich
Qualitätsmanagement- nach DIN EN ISO 13485:2010-01 prozessorientiert Version 0 / Exemplar Nr.: QMH unterliegt dem Änderungsdienst: x Informationsexemplar: Hiermit wird das vorliegende für gültig und verbindlich
EUROPÄISCHE KOMMISSION
 24.1.2013 Amtsblatt der Europäischen Union C 22/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
24.1.2013 Amtsblatt der Europäischen Union C 22/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
Niederspannungsrichtlinie 2014/35/EU Änderungen und Anforderungen. EU-Beratungsstelle der TÜV Rheinland Consulting
 Niederspannungsrichtlinie 2014/35/EU Änderungen und Anforderungen Stefan Rost, 24.11.2015, Leipzig 1 EU-Beratungsstelle der TÜV Rheinland Consulting TÜV Rheinland Consulting GmbH EU-Beratungsstelle Tillystrasse
Niederspannungsrichtlinie 2014/35/EU Änderungen und Anforderungen Stefan Rost, 24.11.2015, Leipzig 1 EU-Beratungsstelle der TÜV Rheinland Consulting TÜV Rheinland Consulting GmbH EU-Beratungsstelle Tillystrasse
Risikoanalyse im Licht der neuen Medizinprodukterichtlinie
 3. Fms Regionalforum, 23. Mai 2008, Leipzig Risikoanalyse im Licht der neuen Medizinprodukterichtlinie Erfahrungen eines Prüfinstituts zu neuer Qualität & Sicherheit im ingenieurtechnischen Team Bildungsangebote
3. Fms Regionalforum, 23. Mai 2008, Leipzig Risikoanalyse im Licht der neuen Medizinprodukterichtlinie Erfahrungen eines Prüfinstituts zu neuer Qualität & Sicherheit im ingenieurtechnischen Team Bildungsangebote
Know-How für die Medizintechnik
 CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
Delta Audit - Fragenkatalog ISO 9001:2014 DIS
 QUMedia GbR Eisenbahnstraße 41 79098 Freiburg Tel. 07 61 / 29286-50 Fax 07 61 / 29286-77 E-mail info@qumedia.de www.qumedia.de Delta Audit - Fragenkatalog ISO 9001:2014 DIS Zur Handhabung des Audit - Fragenkatalogs
QUMedia GbR Eisenbahnstraße 41 79098 Freiburg Tel. 07 61 / 29286-50 Fax 07 61 / 29286-77 E-mail info@qumedia.de www.qumedia.de Delta Audit - Fragenkatalog ISO 9001:2014 DIS Zur Handhabung des Audit - Fragenkatalogs
Risikobasierte Bewertung von Hilfsstoffen
 Risikobasierte Bewertung von Hilfsstoffen Systematische Vorgehensweise beim Risikomanagement-Prozess (in Anlehnung an ICH Q9): Systematische Vorgehensweise beim Risikomanagement-Prozess (in Anlehnung an
Risikobasierte Bewertung von Hilfsstoffen Systematische Vorgehensweise beim Risikomanagement-Prozess (in Anlehnung an ICH Q9): Systematische Vorgehensweise beim Risikomanagement-Prozess (in Anlehnung an
Softwarevalidierung aus Anwendersicht. DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009
 Softwarevalidierung aus Anwendersicht DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009 Softwarevalidierung aus Anwendersicht Geräte mit automatischen Prozessabläufen zur Aufbereitung von Medizinprodukten
Softwarevalidierung aus Anwendersicht DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009 Softwarevalidierung aus Anwendersicht Geräte mit automatischen Prozessabläufen zur Aufbereitung von Medizinprodukten
Rückverfolgbarkeit ISO 13485
 ISO 13485 Normforderung Rückverfolgbarkeit Stolpersteine aus Sicht einer Zertifizierungsstelle ISO 13485 Die Norm ISO 13485: Zweck: Zur Unterstützung der Erfüllung der grundlegenden Anforderungen der europäischen
ISO 13485 Normforderung Rückverfolgbarkeit Stolpersteine aus Sicht einer Zertifizierungsstelle ISO 13485 Die Norm ISO 13485: Zweck: Zur Unterstützung der Erfüllung der grundlegenden Anforderungen der europäischen
Open Source Software als Medizinprodukt
 Open Source Software als Medizinprodukt 1 Begrüßung Innovativer PACS-Hersteller seit 1996 seit 2005 im OsiriX Projekt dabei Marktführer im DICOM Paperprinting 2 Themen Open Source Software (OSS) OsiriX
Open Source Software als Medizinprodukt 1 Begrüßung Innovativer PACS-Hersteller seit 1996 seit 2005 im OsiriX Projekt dabei Marktführer im DICOM Paperprinting 2 Themen Open Source Software (OSS) OsiriX
Dok.-Nr.: Seite 1 von 6
 Logo Apotheke Planung, Durchführung und Dokumentation von QM-Audits Standardarbeitsanweisung (SOP) Standort des Originals: Dok.-Nr.: Seite 1 von 6 Nummer der vorliegenden Verfaßt durch Freigabe durch Apothekenleitung
Logo Apotheke Planung, Durchführung und Dokumentation von QM-Audits Standardarbeitsanweisung (SOP) Standort des Originals: Dok.-Nr.: Seite 1 von 6 Nummer der vorliegenden Verfaßt durch Freigabe durch Apothekenleitung
Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege
 Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege. Dipl. Ing. Norbert Kamps Referent für Hilfsmittelversorgung Medizinischer Dienst des Spitzenverbandes Bund der Krankenkassen e.v. Fachgebiet
Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege. Dipl. Ing. Norbert Kamps Referent für Hilfsmittelversorgung Medizinischer Dienst des Spitzenverbandes Bund der Krankenkassen e.v. Fachgebiet
Medizintechnik und IT
 Medizintechnik und IT Software als Medizinprodukt Alarmierung St.-Marien-Hospital Lünen 10.12.2013 1 MDD 2007/47/EG - Software Software als solche ist ein Medizinprodukt, wenn sie spezifisch vom Hersteller
Medizintechnik und IT Software als Medizinprodukt Alarmierung St.-Marien-Hospital Lünen 10.12.2013 1 MDD 2007/47/EG - Software Software als solche ist ein Medizinprodukt, wenn sie spezifisch vom Hersteller
Qualitätsbeauftragte für Hämotherapie Diskussionsbeitrag aus Erlangen
 Qualitätsbeauftragte für Hämotherapie Diskussionsbeitrag aus Erlangen PD Dr. R. Zimmermann Transfusionsmedizinische und Hämostaseologische Abteilung, Universitätsklinikum Erlangen Transfusionsmedizinische
Qualitätsbeauftragte für Hämotherapie Diskussionsbeitrag aus Erlangen PD Dr. R. Zimmermann Transfusionsmedizinische und Hämostaseologische Abteilung, Universitätsklinikum Erlangen Transfusionsmedizinische
ISO 9001:2015 REVISION. Die neue Struktur mit veränderten Schwerpunkten wurde am 23. September 2015 veröffentlicht und ist seit 15.09.
 ISO 9001:2015 REVISION Die neue Struktur mit veränderten Schwerpunkten wurde am 23. September 2015 veröffentlicht und ist seit 15.09.2015 in Kraft 1 Präsentationsinhalt Teil 1: Gründe und Ziele der Revision,
ISO 9001:2015 REVISION Die neue Struktur mit veränderten Schwerpunkten wurde am 23. September 2015 veröffentlicht und ist seit 15.09.2015 in Kraft 1 Präsentationsinhalt Teil 1: Gründe und Ziele der Revision,
MEDIZINPRODUKTEABGABENVERORDNUNG
 MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
Keine CE-Kennzeichnung ohne klinische Bewertung
 Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Was muss ich noch für meine Zertifizierung tun, wenn meine Organisation. organisiert
 ? organisiert Was muss ich noch für meine Zertifizierung tun, wenn meine Organisation ist? Sie müssen ein QM-System: aufbauen, dokumentieren, verwirklichen, aufrechterhalten und dessen Wirksamkeit ständig
? organisiert Was muss ich noch für meine Zertifizierung tun, wenn meine Organisation ist? Sie müssen ein QM-System: aufbauen, dokumentieren, verwirklichen, aufrechterhalten und dessen Wirksamkeit ständig
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Änderung der ISO/IEC 17025 Anpassung an ISO 9001: 2000
 Änderung der ISO/IEC 17025 Anpassung an ISO 9001: 2000 Dr. Martin Czaske Sitzung der DKD-FA HF & Optik, GS & NF am 11. bzw. 13. Mai 2004 Änderung der ISO/IEC 17025 Anpassung der ISO/IEC 17025 an ISO 9001:
Änderung der ISO/IEC 17025 Anpassung an ISO 9001: 2000 Dr. Martin Czaske Sitzung der DKD-FA HF & Optik, GS & NF am 11. bzw. 13. Mai 2004 Änderung der ISO/IEC 17025 Anpassung der ISO/IEC 17025 an ISO 9001:
Qualitätsmanagement in Gesundheitstelematik und Telemedizin: Sind ISO 9001 basierte Managementsysteme geeignet?
 DGG e.v. PRE-WORKSHOP TELEMED BERLIN 2009 Qualitätsmanagement in Gesundheitstelematik und Telemedizin: Sind ISO 9001 basierte Managementsysteme geeignet? Dr. med. Markus Lindlar Deutsches Zentrum für Luft-
DGG e.v. PRE-WORKSHOP TELEMED BERLIN 2009 Qualitätsmanagement in Gesundheitstelematik und Telemedizin: Sind ISO 9001 basierte Managementsysteme geeignet? Dr. med. Markus Lindlar Deutsches Zentrum für Luft-
Praktische Erfahrungen bei der Kontrolle der Aufbereitung von Medizinprodukten. Andreas Modes
 Praktische Erfahrungen bei der Kontrolle der Aufbereitung von Medizinprodukten Andreas Modes Landesdirektion Dresden Abteilung Arbeitsschutz Mail: andreas.modes@ldd.sachsen.de Tel.: 0375/3903282 Fax: 0375/3903220
Praktische Erfahrungen bei der Kontrolle der Aufbereitung von Medizinprodukten Andreas Modes Landesdirektion Dresden Abteilung Arbeitsschutz Mail: andreas.modes@ldd.sachsen.de Tel.: 0375/3903282 Fax: 0375/3903220
WS 2011 / 2012 Spezielles Arzneimittelrecht Industrielle Arzneimittelherstellung versus Individualherstellung Teil II 2. November 2011 in Mainz
 WS 2011 / 2012 Spezielles Arzneimittelrecht Industrielle Arzneimittelherstellung versus Individualherstellung Teil II in Mainz Folie 1 2 Begriffsbestimmungen 3. ist der EG-GMP Leitfaden (BAnz. S. 6887)
WS 2011 / 2012 Spezielles Arzneimittelrecht Industrielle Arzneimittelherstellung versus Individualherstellung Teil II in Mainz Folie 1 2 Begriffsbestimmungen 3. ist der EG-GMP Leitfaden (BAnz. S. 6887)
Das BASG / AGES PharmMed
 CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
Sicherheitsbewertungsbericht
 Sicherheitsbewertungsbericht auf Basis der "Verordnung (EG) Nr. 352/2009 der Kommission vom 24. April 2009 über die Festlegung einer gemeinsamen Sicherheitsmethode für die Evaluierung und Bewertung von
Sicherheitsbewertungsbericht auf Basis der "Verordnung (EG) Nr. 352/2009 der Kommission vom 24. April 2009 über die Festlegung einer gemeinsamen Sicherheitsmethode für die Evaluierung und Bewertung von
DGQ Regionalkreis Hamburg 21.05.2012 ISO 10007. Konfigurationsmanagement
 DGQ Regionalkreis Hamburg 21.05.2012 ISO 10007 Leitfaden zum Konfigurationsmanagement g Geschichte des Konfigurationsmanagements Mit stetig steigender Produktkomplexität entstanden zunehmend Probleme (z.b.
DGQ Regionalkreis Hamburg 21.05.2012 ISO 10007 Leitfaden zum Konfigurationsmanagement g Geschichte des Konfigurationsmanagements Mit stetig steigender Produktkomplexität entstanden zunehmend Probleme (z.b.
Bereich. Thomas Kauer
 Aktuelle Entwicklungen im PACS- Bereich Thomas Kauer Überblick Aktuelle Entwicklungen im PACS-Bereich Im Tagungsverlauf g Aktuelle Entwicklungen im PACS-Bereich Drei weitere Aspekte Non-DICOM-Bilddatenmanagement
Aktuelle Entwicklungen im PACS- Bereich Thomas Kauer Überblick Aktuelle Entwicklungen im PACS-Bereich Im Tagungsverlauf g Aktuelle Entwicklungen im PACS-Bereich Drei weitere Aspekte Non-DICOM-Bilddatenmanagement
DOT. implantsource. Qualitätsmanagement. Innovative Produkte für die Medizin. Prof. Dr. H.- G.Neumann DOT
 DOT implantsource Qualitätsmanagement Innovative Produkte für die Medizin Prof. Dr. H.- G.Neumann DOT Medizinprodukt - Begriff Medizinprodukte Medizinprodukte nach 3 MPG sind alle einzeln oder miteinander
DOT implantsource Qualitätsmanagement Innovative Produkte für die Medizin Prof. Dr. H.- G.Neumann DOT Medizinprodukt - Begriff Medizinprodukte Medizinprodukte nach 3 MPG sind alle einzeln oder miteinander
Beispielfragen L4(3) Systemauditor nach AS/EN9100 (1st,2nd party)
 Systemauditor nach AS/EN9100 (1st,2nd party)") Allgemeine Hinweise: Es wird von den Teilnehmern erwartet, dass ausreichende Kenntnisse vorhanden sind, um die Fragen 1.1 bis 1.10 unter Verwendung der EN 9100 und ISO 19011 innerhalb von 20 Minuten zu
Allgemeine Hinweise: Es wird von den Teilnehmern erwartet, dass ausreichende Kenntnisse vorhanden sind, um die Fragen 1.1 bis 1.10 unter Verwendung der EN 9100 und ISO 19011 innerhalb von 20 Minuten zu
kundenbezogene Prozesse Produktion und Dienstleistung Lenkung von Überwachungs- und Messmitteln
 Planung Produktrealisierung - Überblick - DIN EN ISO 9001 kundenbezogene Prozesse Entwicklung Beschaffung Produktion und Dienstleistung Lenkung von Überwachungs- und Messmitteln Abb. 1 Qualitätswirksame
Planung Produktrealisierung - Überblick - DIN EN ISO 9001 kundenbezogene Prozesse Entwicklung Beschaffung Produktion und Dienstleistung Lenkung von Überwachungs- und Messmitteln Abb. 1 Qualitätswirksame
Einführung Qualitätsmanagement 2 QM 2
 Einführung Qualitätsmanagement 2 QM 2 Stand: 13.04.2015 Vorlesung 2 Agenda: 1. Reklamationsmanagement (Rekla) 2. Lieferantenbewertung (Lief.bew.) 3. Fehler-Möglichkeits-Einfluss-Analyse (FMEA) 4. Auditmanagement
Einführung Qualitätsmanagement 2 QM 2 Stand: 13.04.2015 Vorlesung 2 Agenda: 1. Reklamationsmanagement (Rekla) 2. Lieferantenbewertung (Lief.bew.) 3. Fehler-Möglichkeits-Einfluss-Analyse (FMEA) 4. Auditmanagement
Anhang V EG-Konformitätserklärung (Qualitätssicherung Produktion)
") Dieses Werk, einschließlich aller seiner Teile, ist urheberrechtlich geschützt. Jede Verwertung außerhalb der engen Grenzen des Urheberrechtsgesetzes ist ohne Zustimmung des Verlages unzulässig und strafbar.
Dieses Werk, einschließlich aller seiner Teile, ist urheberrechtlich geschützt. Jede Verwertung außerhalb der engen Grenzen des Urheberrechtsgesetzes ist ohne Zustimmung des Verlages unzulässig und strafbar.
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Dieter Brunner ISO 27001 in der betrieblichen Praxis
 Seite 1 von 6 IT-Sicherheit: die traditionellen Sichtweise Traditionell wird Computer-Sicherheit als technisches Problem gesehen Technik kann Sicherheitsprobleme lösen Datenverschlüsselung, Firewalls,
Seite 1 von 6 IT-Sicherheit: die traditionellen Sichtweise Traditionell wird Computer-Sicherheit als technisches Problem gesehen Technik kann Sicherheitsprobleme lösen Datenverschlüsselung, Firewalls,
Einführung Risk Management Konzept
 Einführung Risk Management Konzept 1. Risiko unser ständiger Begleiter Das Risk Management ist ein wichtiges Führungsinstrument für das Erreichen der Zielsetzungen und für den Schutz der Mitarbeitenden,
Einführung Risk Management Konzept 1. Risiko unser ständiger Begleiter Das Risk Management ist ein wichtiges Führungsinstrument für das Erreichen der Zielsetzungen und für den Schutz der Mitarbeitenden,
(Veröffentlichung der Titel und der Bezugsnummern der harmonisierten Normen im Sinne der Harmonisierungsrechtsvorschriften
 C 14/36 DE Amtsblatt der Europäischen Union 16.1.2015 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschriften der
C 14/36 DE Amtsblatt der Europäischen Union 16.1.2015 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschriften der
Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium
 QUALITY-APPS Applikationen für das Qualitätsmanagement Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium Autor: Prof. Dr. Jürgen P. Bläsing Die Maschinenrichtlinie 2006/42/EG ist
QUALITY-APPS Applikationen für das Qualitätsmanagement Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium Autor: Prof. Dr. Jürgen P. Bläsing Die Maschinenrichtlinie 2006/42/EG ist
für die Aufbereitung von Medizinprodukten
 Die Bedeutung der Biokompatibilität für die Aufbereitung von Medizinprodukten Dr.-Ing. Ute Müller, Geschäftsführer BMP Labor für medizinische Materialprüfung GmbH, Aachen Inhalt Einordnung der Biokompatibilität
Die Bedeutung der Biokompatibilität für die Aufbereitung von Medizinprodukten Dr.-Ing. Ute Müller, Geschäftsführer BMP Labor für medizinische Materialprüfung GmbH, Aachen Inhalt Einordnung der Biokompatibilität
1.1. Aufschriften auf der Außenseite von ME-Geräten oder ME-Geräte-Teilen
 1. Bezeichnung, Aufschriften und Begleitpapiere 1.1. Aufschriften auf der Außenseite von ME-Geräten oder ME-Geräte-Teilen 1.1.1. Aufschriften auf der Außenseite von ME-Geräten oder ME-Geräte-Teilen, die
1. Bezeichnung, Aufschriften und Begleitpapiere 1.1. Aufschriften auf der Außenseite von ME-Geräten oder ME-Geräte-Teilen 1.1.1. Aufschriften auf der Außenseite von ME-Geräten oder ME-Geräte-Teilen, die
Neuerungen und praktische Auswirkungen auf das Qualitäts- und Hygienemanagement durch IFS / Version 5.
 Neuerungen und praktische Auswirkungen auf das Qualitäts- und Hygienemanagement durch IFS / Version 5. Referent: Dipl. Ing. H. Klein Fleischtechnologe QM - Beratung, Bünde Ziel des IFS Nachweis eines funktionierenden
Neuerungen und praktische Auswirkungen auf das Qualitäts- und Hygienemanagement durch IFS / Version 5. Referent: Dipl. Ing. H. Klein Fleischtechnologe QM - Beratung, Bünde Ziel des IFS Nachweis eines funktionierenden
1.1. Geltungsbereich Diese Prüfungsordnung gilt für die Prüfung zum Technischen Risikomanager nach DIN VDE V 0827.
 www.dgwz.de/risikomanager Prüfungsordnung für die Prüfung zum Technischen Risikomanager nach DIN VDE V 0827 DGWZ 1013:2015-07 Stand: Juli 2015 Inhaltsverzeichnis 1. Allgemeines... 1 1.1. Geltungsbereich...
www.dgwz.de/risikomanager Prüfungsordnung für die Prüfung zum Technischen Risikomanager nach DIN VDE V 0827 DGWZ 1013:2015-07 Stand: Juli 2015 Inhaltsverzeichnis 1. Allgemeines... 1 1.1. Geltungsbereich...
Information zur Revision der ISO 9001. Sehr geehrte Damen und Herren,
 Sehr geehrte Damen und Herren, mit diesem Dokument möchten wir Sie über die anstehende Revision der ISO 9001 und die sich auf die Zertifizierung ergebenden Auswirkungen informieren. Die folgenden Informationen
Sehr geehrte Damen und Herren, mit diesem Dokument möchten wir Sie über die anstehende Revision der ISO 9001 und die sich auf die Zertifizierung ergebenden Auswirkungen informieren. Die folgenden Informationen
Einführung: Der erfolgreiche Unsinn mit der ISO 9001...1 1. Grundsatzfragen zur Darlegung...6
 Inhaltsverzeichnis Einführung: Der erfolgreiche Unsinn mit der ISO 9001...1 1. Grundsatzfragen zur Darlegung...6 1.1 Genormte Ungereimtheiten...6 1.1.1 Das Qualitätsmanagementsystem einführen?...6 1.1.2
Inhaltsverzeichnis Einführung: Der erfolgreiche Unsinn mit der ISO 9001...1 1. Grundsatzfragen zur Darlegung...6 1.1 Genormte Ungereimtheiten...6 1.1.1 Das Qualitätsmanagementsystem einführen?...6 1.1.2
Datenschutz-Management
 Dienstleistungen Datenschutz-Management Datenschutz-Management Auf dem Gebiet des Datenschutzes lauern viele Gefahren, die ein einzelnes Unternehmen oft nur schwer oder erst spät erkennen kann. Deshalb
Dienstleistungen Datenschutz-Management Datenschutz-Management Auf dem Gebiet des Datenschutzes lauern viele Gefahren, die ein einzelnes Unternehmen oft nur schwer oder erst spät erkennen kann. Deshalb
Übungsbeispiele für die mündliche Prüfung
 Übungsbeispiele für die mündliche Prüfung Nr. Frage: 71-02m Welche Verantwortung und Befugnis hat der Beauftragte der Leitung? 5.5.2 Leitungsmitglied; sicherstellen, dass die für das Qualitätsmanagementsystem
Übungsbeispiele für die mündliche Prüfung Nr. Frage: 71-02m Welche Verantwortung und Befugnis hat der Beauftragte der Leitung? 5.5.2 Leitungsmitglied; sicherstellen, dass die für das Qualitätsmanagementsystem
9001 weitere (kleinere) Änderungen
 Änderungen") 6.2 Ziele: SMARTE Ziele: was, Ressorucen, Verantwortung, Termin, Bewertung der Ergebnisse (für ecco nicht nue, wurde aber betont) 6.3 Änderungen: Der Einfluss von Änderungen am QMS uss bewertet werden
6.2 Ziele: SMARTE Ziele: was, Ressorucen, Verantwortung, Termin, Bewertung der Ergebnisse (für ecco nicht nue, wurde aber betont) 6.3 Änderungen: Der Einfluss von Änderungen am QMS uss bewertet werden
SAFEYTEAMS-Newsletter Nr. 5
 CE-Kennzeichnung I Gefahrenanalysen I Maschinen-Prüfungen I Workshops I Seminare SAFEYTEAMS-Newsletter Nr. 5 Thema Bedeutung des Performance-Levels (PL) Definition nach Norm EN 13849: Diskreter Level,
CE-Kennzeichnung I Gefahrenanalysen I Maschinen-Prüfungen I Workshops I Seminare SAFEYTEAMS-Newsletter Nr. 5 Thema Bedeutung des Performance-Levels (PL) Definition nach Norm EN 13849: Diskreter Level,
Prozessbewertung und -verbesserung nach ITIL im Kontext des betrieblichen Informationsmanagements. von Stephanie Wilke am 14.08.08
 Prozessbewertung und -verbesserung nach ITIL im Kontext des betrieblichen Informationsmanagements von Stephanie Wilke am 14.08.08 Überblick Einleitung Was ist ITIL? Gegenüberstellung der Prozesse Neuer
Prozessbewertung und -verbesserung nach ITIL im Kontext des betrieblichen Informationsmanagements von Stephanie Wilke am 14.08.08 Überblick Einleitung Was ist ITIL? Gegenüberstellung der Prozesse Neuer
Gründe für fehlende Vorsorgemaßnahmen gegen Krankheit
 Gründe für fehlende Vorsorgemaßnahmen gegen Krankheit politische Lage verlassen sich auf Familie persönliche, finanzielle Lage meinen, sich Vorsorge leisten zu können meinen, sie seien zu alt nicht mit
Gründe für fehlende Vorsorgemaßnahmen gegen Krankheit politische Lage verlassen sich auf Familie persönliche, finanzielle Lage meinen, sich Vorsorge leisten zu können meinen, sie seien zu alt nicht mit
8.5. Medizinproduktegesetz
 8.5. Medizinproduktegesetz 1 Zweck des Gesetzes http://bundesrecht.juris.de/bundesrecht/mpg/ 3 Begriffsbestimmungen Zweck dieses Gesetzes ist es, den Verkehr mit Medizinprodukten zu regeln und dadurch
8.5. Medizinproduktegesetz 1 Zweck des Gesetzes http://bundesrecht.juris.de/bundesrecht/mpg/ 3 Begriffsbestimmungen Zweck dieses Gesetzes ist es, den Verkehr mit Medizinprodukten zu regeln und dadurch
PLM: Privat Label Manufacturer (Kunde der OEM-PLM-Beziehung) OEM: Original Equipment Manufacturer (Lieferant der OEM-PLM-Beziehung)
 OEM: Original Equipment Manufacturer (Lieferant der OEM-PLM-Beziehung)") 1 Anwendsbereich Prüf der Produktdokumentation im Falle von Privat Label Herstellern gemäß OEM Verfahren Zu verwenden bei neuen Anträgen zur Zertifizier und bei Stichprobenprüfen im Rahmen der regelmäßigen
1 Anwendsbereich Prüf der Produktdokumentation im Falle von Privat Label Herstellern gemäß OEM Verfahren Zu verwenden bei neuen Anträgen zur Zertifizier und bei Stichprobenprüfen im Rahmen der regelmäßigen
Agile Vorgehensmodelle in der Softwareentwicklung: Scrum
 C A R L V O N O S S I E T Z K Y Agile Vorgehensmodelle in der Softwareentwicklung: Scrum Johannes Diemke Vortrag im Rahmen der Projektgruppe Oldenburger Robot Soccer Team im Wintersemester 2009/2010 Was
C A R L V O N O S S I E T Z K Y Agile Vorgehensmodelle in der Softwareentwicklung: Scrum Johannes Diemke Vortrag im Rahmen der Projektgruppe Oldenburger Robot Soccer Team im Wintersemester 2009/2010 Was
PRÜFMODUL D UND CD. 1 Zweck. 2 Durchführung. 2.1 Allgemeines. 2.2 Antrag
 1 Zweck PRÜFMODUL D UND CD Diese Anweisung dient als Basis für unsere Kunden zur Information des Ablaufes der folgenden EG-Prüfung nach folgenden Prüfmodulen: D CD Es beschreibt die Aufgabe der benannten
1 Zweck PRÜFMODUL D UND CD Diese Anweisung dient als Basis für unsere Kunden zur Information des Ablaufes der folgenden EG-Prüfung nach folgenden Prüfmodulen: D CD Es beschreibt die Aufgabe der benannten
GPP Projekte gemeinsam zum Erfolg führen
 GPP Projekte gemeinsam zum Erfolg führen IT-Sicherheit Schaffen Sie dauerhaft wirksame IT-Sicherheit nach zivilen oder militärischen Standards wie der ISO 27001, dem BSI Grundschutz oder der ZDv 54/100.
GPP Projekte gemeinsam zum Erfolg führen IT-Sicherheit Schaffen Sie dauerhaft wirksame IT-Sicherheit nach zivilen oder militärischen Standards wie der ISO 27001, dem BSI Grundschutz oder der ZDv 54/100.
ippl uality anagement begrüßt Sie herzlich zum heutigen Informationsabend 14.09.09 Qualitätsmanagement ISO 9001 1
 begrüßt Sie herzlich zum heutigen Informationsabend Qualitätsmanagement ISO 9001 1 Wer aufhört besser zu werden, hat aufgehört gut zu sein! (Philip Rosenthal) Qualitätsmanagement ISO 9001 2 QUALITÄT und
begrüßt Sie herzlich zum heutigen Informationsabend Qualitätsmanagement ISO 9001 1 Wer aufhört besser zu werden, hat aufgehört gut zu sein! (Philip Rosenthal) Qualitätsmanagement ISO 9001 2 QUALITÄT und
Pensionskasse des Bundes Caisse fédérale de pensions Holzikofenweg 36 Cassa pensioni della Confederazione
 Compliance-Reglement 1. Grundsätze und Ziele Compliance ist die Summe aller Strukturen und Prozesse, die sicherstellen, dass und ihre Vertreter/Vertreterinnen alle relevanten Gesetze, Vorschriften, Codes
Compliance-Reglement 1. Grundsätze und Ziele Compliance ist die Summe aller Strukturen und Prozesse, die sicherstellen, dass und ihre Vertreter/Vertreterinnen alle relevanten Gesetze, Vorschriften, Codes
Qualitäts- Managementhandbuch (QMH) DIN EN ISO 9001 : 2008 (ohne Entwicklung) von. Margot Schön Burgbühl 11 88074 Meckenbeuren
 DIN EN ISO 9001 : 2008 (ohne Entwicklung) von. Margot Schön Burgbühl 11 88074 Meckenbeuren") Qualitäts- Managementhandbuch (QMH) DIN EN ISO 9001 : 2008 (ohne Entwicklung) von Margot Schön Die Einsicht Führung des IMS-Handbuches ist EDV-technisch verwirklicht. Jeder Ausdruck unterliegt nicht dem
Qualitäts- Managementhandbuch (QMH) DIN EN ISO 9001 : 2008 (ohne Entwicklung) von Margot Schön Die Einsicht Führung des IMS-Handbuches ist EDV-technisch verwirklicht. Jeder Ausdruck unterliegt nicht dem
9.6 Korrekturmaßnahmen, Qualitätsverbesserung
 Teil III Organisation und Infrastruktur Kapitel 9: Qualitätsmanagementsystem Inhalt 9.1 Grundlagen 9.2 Qualitätspolitik 9.3 Qualitätsorganisation 9.4 Maßnahmen 9.5 Qualitätsaufzeichnungen 9.6 Korrekturmaßnahmen,
Teil III Organisation und Infrastruktur Kapitel 9: Qualitätsmanagementsystem Inhalt 9.1 Grundlagen 9.2 Qualitätspolitik 9.3 Qualitätsorganisation 9.4 Maßnahmen 9.5 Qualitätsaufzeichnungen 9.6 Korrekturmaßnahmen,
Dokumentenlenkung - Pflicht oder Kür-
 Dokumentenlenkung - Pflicht oder Kür- - QM-Sprengel Württemberg - Sunhild Klöss Stabsabteilung Projekt- und Qualitätsmanagement Klinikum Heidenheim Themenübersicht Forderungen der DIN EN ISO 9001 Was muss
Dokumentenlenkung - Pflicht oder Kür- - QM-Sprengel Württemberg - Sunhild Klöss Stabsabteilung Projekt- und Qualitätsmanagement Klinikum Heidenheim Themenübersicht Forderungen der DIN EN ISO 9001 Was muss
experttyjverlag Das QM-System nach DIN EN ISO 9001 und DIN EN ISO 13485 für Medizinprodukte. Hilfen zur Darlegung und zum Risikomanagement
 Das QM-System nach DIN EN ISO 9001 und DIN EN ISO 13485 für Medizinprodukte. Hilfen zur Darlegung und zum Risikomanagement Hinrich Franke 3., völlig neu bearbeitete und erweiterte Auflage experttyjverlag
Das QM-System nach DIN EN ISO 9001 und DIN EN ISO 13485 für Medizinprodukte. Hilfen zur Darlegung und zum Risikomanagement Hinrich Franke 3., völlig neu bearbeitete und erweiterte Auflage experttyjverlag
wir müssen reden. Über Qualität!
 wir müssen reden. Über Qualität! "Made in Quality - Made for Success" 1 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack Warum Auditors Liebling? Es ist eine muss Forderung jeder Systemnorm!
wir müssen reden. Über Qualität! "Made in Quality - Made for Success" 1 Auditors Liebling! Der Messmittelmanagementprozess Jörg Roggensack Warum Auditors Liebling? Es ist eine muss Forderung jeder Systemnorm!
Das Pflichtenheft. Dipl.- Ing. Dipl.-Informatiker Dieter Klapproth Ains A-Systemhaus GmbH Berlin
 Fragestellungen: Warum reicht das Lastenheft nicht aus? Was kann ich mit dem Lastenheft machen? Was unterscheidet das Pflichtenheft vom Lastenheft? Was gehört zum Auftragsumfang einer Individualsoftware?
Fragestellungen: Warum reicht das Lastenheft nicht aus? Was kann ich mit dem Lastenheft machen? Was unterscheidet das Pflichtenheft vom Lastenheft? Was gehört zum Auftragsumfang einer Individualsoftware?
Qualitätsmanagement wie es im Buche steht
 Qualitätsmanagement wie es im Buche steht Ein Praxisbericht aus der Bibliothek der Technischen Universität München Fortbildungsveranstaltung des VdB Regionalverband Südwest und Landesverband Bayern und
Qualitätsmanagement wie es im Buche steht Ein Praxisbericht aus der Bibliothek der Technischen Universität München Fortbildungsveranstaltung des VdB Regionalverband Südwest und Landesverband Bayern und
Risikomanagement-Prozess nach EN ISO 14971:2007 Anwendung der FMEA-Systematik
 Risikomanagement-Prozess nach EN ISO 14971:2007 Anwendung der FMEA-Systematik TecPart Fachgruppe Medizintechnik 03.02.2009 1 Inhalt Grundlagen Risikomanagementprozess Wie kann die FMEA-Methodik das Risikomanagement
Risikomanagement-Prozess nach EN ISO 14971:2007 Anwendung der FMEA-Systematik TecPart Fachgruppe Medizintechnik 03.02.2009 1 Inhalt Grundlagen Risikomanagementprozess Wie kann die FMEA-Methodik das Risikomanagement
Anleitung zur Umsetzung der Forderungen der Revision der ISO 9001:2015
 Anleitung zur Umsetzung der Forderungen der Revision der ISO 9001:2015 Änderungen bezüglich Struktur, Terminologie und Konzepte Struktur und Terminologie Die Gliederung (d. h. Abschnittsreihenfolge) und
Anleitung zur Umsetzung der Forderungen der Revision der ISO 9001:2015 Änderungen bezüglich Struktur, Terminologie und Konzepte Struktur und Terminologie Die Gliederung (d. h. Abschnittsreihenfolge) und
Technischer Hinweis Merkblatt DVGW G 1001 (M) März 2015
 März 2015") www.dvgw-regelwerk.de Technischer Hinweis Merkblatt DVGW G 1001 (M) März 2015 Sicherheit in der Gasversorgung; Risikomanagement von gastechnischen Infrastrukturen im Normalbetrieb Security of Gas Supply;
www.dvgw-regelwerk.de Technischer Hinweis Merkblatt DVGW G 1001 (M) März 2015 Sicherheit in der Gasversorgung; Risikomanagement von gastechnischen Infrastrukturen im Normalbetrieb Security of Gas Supply;
Muster Nachweisdokumentation und Sicherheitsbewertungsbericht
 Muster Nachweisdokumentation und Sicherheitsbewertungsbericht auf Basis der "Verordnung (EG) Nr. 352/2009 der Kommission vom 24. April 2009 über die Festlegung einer gemeinsamen Sicherheitsmethode für
Muster Nachweisdokumentation und Sicherheitsbewertungsbericht auf Basis der "Verordnung (EG) Nr. 352/2009 der Kommission vom 24. April 2009 über die Festlegung einer gemeinsamen Sicherheitsmethode für
Betreiben von Aufzugsanlagen nach der Betriebssicherheitsverordnung (BetrSichV)
") Betreiben von Aufzugsanlagen nach der Betriebssicherheitsverordnung (BetrSichV) Europäische Anforderungen an den sicheren Betrieb von Aufzugsanlagen und die Umsetzung in deutsche Gesetzgebung Mai 04 Dipl.-Ing.
Betreiben von Aufzugsanlagen nach der Betriebssicherheitsverordnung (BetrSichV) Europäische Anforderungen an den sicheren Betrieb von Aufzugsanlagen und die Umsetzung in deutsche Gesetzgebung Mai 04 Dipl.-Ing.
Abweichungen. Anforderungen / Zitate aus den Rechtsvorschriften
 Abweichungen Anforderungen / Zitate aus den Rechtsvorschriften AMWHV [...] Alle Abweichungen im Prozess und von der Festlegung der Spezifikation sind zu dokumentieren und gründlich zu untersuchen. [...]
Abweichungen Anforderungen / Zitate aus den Rechtsvorschriften AMWHV [...] Alle Abweichungen im Prozess und von der Festlegung der Spezifikation sind zu dokumentieren und gründlich zu untersuchen. [...]
Die insoweit erfahrene Fachkraft Gemäß 8a, Abs. 2 SGB VIII
 Die insoweit erfahrene Fachkraft Gemäß 8a, Abs. 2 SGB VIII Verortung Qualifikation Aufgaben Lotte Knoller, Diplom Psychologin, Kinderschutz-Zentrum Berlin 8a Schutzauftrag bei Kindeswohlgefährdung (1)
Die insoweit erfahrene Fachkraft Gemäß 8a, Abs. 2 SGB VIII Verortung Qualifikation Aufgaben Lotte Knoller, Diplom Psychologin, Kinderschutz-Zentrum Berlin 8a Schutzauftrag bei Kindeswohlgefährdung (1)
Prüfmittelmanagement
 W. Kistner Prüfmittelmanagement Prüfmittelmanagement Nutzen und Bedeutung in der industriellen Praxis Vorstellung des neuen DGQ-Bandes 13-61 Prüfmittelmanagement Wolfgang Kistner, Kistner Meßtechnik Obmann
W. Kistner Prüfmittelmanagement Prüfmittelmanagement Nutzen und Bedeutung in der industriellen Praxis Vorstellung des neuen DGQ-Bandes 13-61 Prüfmittelmanagement Wolfgang Kistner, Kistner Meßtechnik Obmann
Themenarbeit HTA.SWE.S08 Pascal Ming 23.Juni 2008
 Themenarbeit HTA.SWE.S08 Pascal Ming 23.Juni 2008 Einleitung Risikomanagement nach HTAgil Risikomanagement nach Bärentango Risikomanagement in Wikipedia Vergleich Aufgabe Risikomanagement(Jörg Hofstetter)
Themenarbeit HTA.SWE.S08 Pascal Ming 23.Juni 2008 Einleitung Risikomanagement nach HTAgil Risikomanagement nach Bärentango Risikomanagement in Wikipedia Vergleich Aufgabe Risikomanagement(Jörg Hofstetter)
5 JAHRE EN ISO 13485 bereits Routine?
 5 JAHRE EN ISO 13485 bereits Routine? Erfahrungsbericht aus einer zertifizierten AEMP Brigitte Keplinger Krankenhaus Barmherzige Schwestern Linz Akutkrankenhaus mit Schwerpunkt Onkologie Kardiologie Orthopädie
5 JAHRE EN ISO 13485 bereits Routine? Erfahrungsbericht aus einer zertifizierten AEMP Brigitte Keplinger Krankenhaus Barmherzige Schwestern Linz Akutkrankenhaus mit Schwerpunkt Onkologie Kardiologie Orthopädie
Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203
 TRBS 1111 TRBS 2121 TRBS 1203") Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203 Achim Eckert 1/12 Am 3. Oktober 2002 ist die Betriebssicherheitsverordnung in Kraft getreten. Auch für den Gerüstbauer und den
Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203 Achim Eckert 1/12 Am 3. Oktober 2002 ist die Betriebssicherheitsverordnung in Kraft getreten. Auch für den Gerüstbauer und den
Ablauf Vorstellungsgespräch
 Leitfaden für Vorstellungsgespräche Ablauf Vorstellungsgespräch Bewerber: Bewerbung als: Interviewer: Datum: ERGEBNIS DES VORSTELLUNGSGESPRÄCHS Gesamtpunktzahl 14-16 Hervorragend 9 13 Kompetent 6-8 Entwicklungsbedarf
Leitfaden für Vorstellungsgespräche Ablauf Vorstellungsgespräch Bewerber: Bewerbung als: Interviewer: Datum: ERGEBNIS DES VORSTELLUNGSGESPRÄCHS Gesamtpunktzahl 14-16 Hervorragend 9 13 Kompetent 6-8 Entwicklungsbedarf
Ziel- und Qualitätsorientierung. Fortbildung für die Begutachtung in Verbindung mit dem Gesamtplanverfahren nach 58 SGB XII
 Ziel- und Qualitätsorientierung Fortbildung für die Begutachtung in Verbindung mit dem Gesamtplanverfahren nach 58 SGB XII Qualität? In der Alltagssprache ist Qualität oft ein Ausdruck für die Güte einer
Ziel- und Qualitätsorientierung Fortbildung für die Begutachtung in Verbindung mit dem Gesamtplanverfahren nach 58 SGB XII Qualität? In der Alltagssprache ist Qualität oft ein Ausdruck für die Güte einer
Risikomanagement in der Praxis Alles Compliance oder was?! 1. IT-Grundschutz-Tag 2014 13.02.2014
 Risikomanagement in der Praxis Alles Compliance oder was?! 1. IT-Grundschutz-Tag 2014 13.02.2014 Risikomanagement Eine Einführung Risikomanagement ist nach der Norm ISO 31000 eine identifiziert, analysiert
Risikomanagement in der Praxis Alles Compliance oder was?! 1. IT-Grundschutz-Tag 2014 13.02.2014 Risikomanagement Eine Einführung Risikomanagement ist nach der Norm ISO 31000 eine identifiziert, analysiert
Der Schutz von Patientendaten
 Der Schutz von Patientendaten bei (vernetzten) Software-Medizinprodukten aus Herstellersicht 18.09.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
Der Schutz von Patientendaten bei (vernetzten) Software-Medizinprodukten aus Herstellersicht 18.09.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
Umsetzung der Hygienerichtlinien
 Umsetzung der Hygienerichtlinien Andrea Percht, MBA Hygienefachkraft allgemein beeidete und gerichtlich zertifizierte Sachverständige für f r Hygiene Richtlinien Diese Leitlinie wurde auf Basis des Medizinproduktegesetzes
Umsetzung der Hygienerichtlinien Andrea Percht, MBA Hygienefachkraft allgemein beeidete und gerichtlich zertifizierte Sachverständige für f r Hygiene Richtlinien Diese Leitlinie wurde auf Basis des Medizinproduktegesetzes
WAS finde ich WO im Beipackzettel
 WAS finde ich WO im Beipackzettel Sie haben eine Frage zu Ihrem? Meist finden Sie die Antwort im Beipackzettel (offiziell "Gebrauchsinformation" genannt). Der Aufbau der Beipackzettel ist von den Behörden
WAS finde ich WO im Beipackzettel Sie haben eine Frage zu Ihrem? Meist finden Sie die Antwort im Beipackzettel (offiziell "Gebrauchsinformation" genannt). Der Aufbau der Beipackzettel ist von den Behörden
Aufbereitung von Medizinprodukten
 R K I - R I C H T L I N I E N Aufbereitung von Medizinprodukten Erläuterungen zu der Übersicht Nachfolgend veröffentlichen wir eine Übersicht zur Aufbereitung von Medizinprodukten der Gruppen semikritisch
R K I - R I C H T L I N I E N Aufbereitung von Medizinprodukten Erläuterungen zu der Übersicht Nachfolgend veröffentlichen wir eine Übersicht zur Aufbereitung von Medizinprodukten der Gruppen semikritisch