Bioinformatik für Lebenswissenschaftler
|
|
|
- Damian Hofmeister
- vor 6 Jahren
- Abrufe
Transkript
1 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS Hiden Markov Models & Phylogenien Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen
2 Übersicht Hidden Markov Model (HMM) Allgemeines Konzept Profile HMM Anwendung Phylogenie Evolutionsmodelle Konstruktion von Stammbäumen Darstellung 2
3 Hidden Markov Models (HMM) HMM sind ein stochastisches Modell, das eine System (z.b. Alignment) als eine Kette von Zufallsprozessen darstellt Sie werden häufig in der Bioinformatik verwendet Identifizierung von kodierenden Regionen oder CpG Sequenz Suche (Profil HMM) Sekundär-Struktur-Vorhersage
 und Übergänge (t(s x,s y )) Jeder")
 mit den jeweiligen")
 zwischen Zuständen ist eine")
=1 S x t(s x,s y )=1 S x t(s 1,S 2 ) t(s 2,S 3 )")
4 HMM - Definition Ein HMM besitzt sogenannte Zustände (S x ) und Übergänge (t(s x,s y )) Jeder Zustand hat eine bestimmte Anzahl von Beobachtungen (E a emission) mit den jeweiligen Wahrscheinlichkeiten p(e a ) Jedem Übergang (t transition) zwischen Zuständen ist eine Wahrscheinlichkeit zugeordnet t(s 1,S 3 ) p(e a )=1 S x t(s x,s y )=1 S x t(s 1,S 2 ) t(s 2,S 3 ) S 1 S 2 S 3 t(s 2,S 1 ) E 1 E 2 E 3 E 4
5 Profil HMM - Definition Es gibt Zusätzlich ein Anfangs- und Endzustand Jeder Pfad durch das Model vom Anfangs- bis zum Endzustand ergibt eine Sequenz Die Beobachtungen (Emissionen) sind in diesem Falle eine As einer Sequenz MSA können als sogenannte Profil HMM dargestellt werden




")
 D i D")
6 Profil HMM - Architektur Jede Spalte im Alignment lässt sich durch 3 Zustände darstellen Match (M) Deletion (D) Insertion (I) D i D i+1 M i M i+1 I i Ii+1


7 Profil HMM - Beispiel Start Stop E E 2 A C D E F G H I K L M N P Q R S T V W Y E E 4 A C D E F G H I K L M N P Q R S T V W Y A C D E F G H I K L M N P Q R S T V W Y A C D E F G H I K L M N P Q R S T V W Y PATH ist die wahrscheinlichste Sequenz, aber auch PETS ist möglich

8 Profil HMM lokale Alignments Insertionen vor und nach HMM werden erlaubt Auch teilweise Alignments sind möglich Start Start Still Still
9 Profil HMM Repeats Das HMM darf mehrmals vorkommen Start Start Still Still
10 HMM aus MSA generieren Die einzelnen Spalten im Alignment müssen den Zuständen zugeordnet werden Match: Spalten ohne gaps Insertion: Spalten mit "vielen" gaps Somit lassen sich die Sequenzen als Pfad im HMM verfolgen Die Gesamtwahrscheinlichkeit ist das Produkt der Wahrscheinlichkeiten für Übergängen und den Beobachtungen
11 Profil HMM - Anwendungen HMMER3 ist eine frei verfügbare Implementation BLASTP / PSI-BLAST ähnliche Suche (phmmer, jackhmmer) Ein HMM aus einem MSA erzeugen (hmmbuild) Consensus oder eine Anzahl von Sequenzen aus einem HMM erzeugen (hmmemit) HMM Suche gegen eine Sequenzdatenbank (hmmsearch) Ein MSA aus Sequenzen erzeugen mit Hilfe eines HMM (hmmalign) Sequenzsuche gegen eine Profil HMM Datenbank (SMART, PFAM, hmmscan) HMM gegen HMM Datenbank (HHPred Söding et al.)
12 Profil HMM vs PSSM Genauso wie PSSMs sind Profil HMM stark abhängig von der Qualität des MSA HMM enthalten Wahrscheinlichkeiten für Insertionen und Deletionen, PSSM (PSI-BLAST) nicht HMM sind deutlich sensitiver, aber auch Rechenzeit Intensiver
13 Vergleich von Suchmethoden


14 Taxonomie Evolution Phylogenie Carl von Linné schlägt um 1735 eine hierarchische Systematik zur Gliederung der Arten vor



15 Taxonomie Evolution Phylogenie Charles Darwins Evolutionstheorie gibt ca. 120 Jahre später eine Erklärung für die Entstehung neuer Arten, die zu baumartigen Hierarchien führt
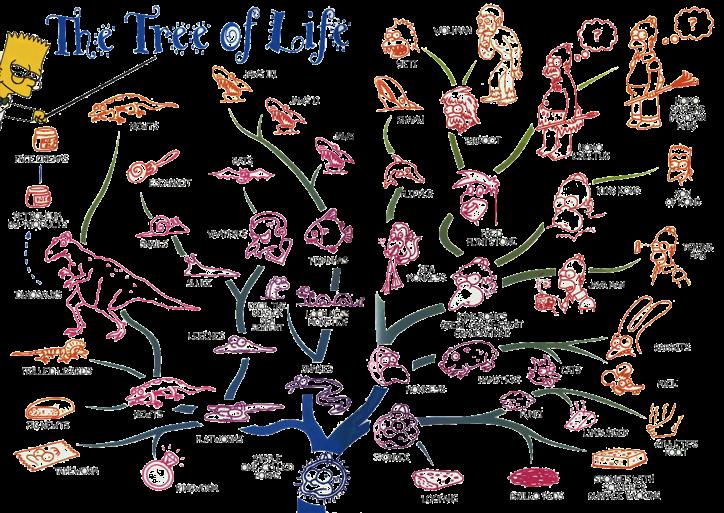
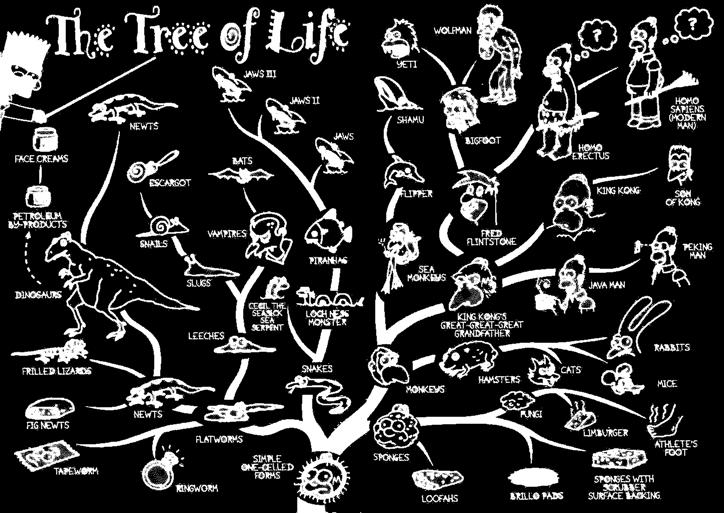
16
17 Taxonomie Evolution - Phylogenie Phylogenie oder Phylogenese beschreibt die evolutionäre Entstehung der Arten Diese Entwicklung kann in Form eines phylogenetischen Baums dargestellt werden Zur Rekonstruktion phylogenetischer Bäume lassen sich verschiedene Methoden heranziehen Morphologischer oder anatomischer Vergleich rezenter Lebewesen Morphologischer oder anatomischer Vergleich fossiler Lebewesen Sequenzanalyse Verwendung der Sequenzanalyse basiert dabei auf der Annahme einer molekularen Uhr


18 Die Molekulare Uhr
19 Die Molekulare Uhr Mutationsraten schwanken stark von Art zu Art, von Gen zu Gen, von Lokus zu Lokus Starker Unterschied zwischen kodierenden und nicht kodierenden Regionen Mitochondriale DNA hat höhere Mutationsraten (Fehlende Korrekturmechanismen)
20 Graphen und Bäume Graphen sind ein wichtiges Konzept in der Informatik Mit Graphen lassen sich viele Alltagsprobleme anschaulich darstellen Man hat umfangreiche mathematische Werkzeuge um abstrakt damit zu arbeiten Kante Ein Graph besteht aus Knoten Kanten (die Knoten miteinander verbinden) Knoten
21 Graphen und Bäume Graphen sind ein wichtiges Konzept in der Informatik Mit Graphen lassen sich viele Alltagsprobleme anschaulich darstellen Man hat umfangreiche mathematische Werkzeuge um abstrakt damit zu arbeiten Ein Graph besteht aus Knoten Kanten (die Knoten miteinander verbinden) Dieser einfache Graph drückt Nachbarschaftsbeziehungen in Europa aus. Zwei Knoten sind durch eine Kante verbunden, wenn die entsprechenden Länder aneinander grenzen.
22 Graphen und Bäume Graphen sind ein wichtiges Konzept in der Informatik Mit Graphen lassen sich viele Alltagsprobleme anschaulich darstellen Man hat umfangreiche mathematische Werkzeuge um abstrakt damit zu arbeiten Ein Graph besteht aus Knoten Kanten (die Knoten miteinander verbinden) H H H C C Dieser Graph stellt die Struktur von Pyridin dar. Knoten stehen für Atome und sind mit dem Elementsymbol beschriftet. zwei Knoten sind durch eine Kante verbunden, wenn die Atome eine Bindung teilen. C N C C H H
23 Graphen und Bäume Es gibt verschiedene Arten von Graphen, die unterschiedliche Eigenschaften haben Graphen können z.b. Zyklen besitzen, d.h. man kann entlang der Kanten von einem Knoten zu sich selbst wandern, ohne eine Kante zweimal zu nutzen
24 Graphen und Bäume Es gibt verschiedene Arten von Graphen, die unterschiedliche Eigenschaften haben Graphen können z.b. Zyklen besitzen, d.h. man kann entlang der Kanten von einem Knoten zu sich selbst wandern, ohne eine Kante zweimal zu nutzen Graphen ohne Zyklen (azyklische Graphen), werden auch Bäume genannt Zyklischer Graph Azyklischer Graph (Baum)
25 Bäume Einfache evolutionäre Beziehungen lassen sich mit Hilfe von Bäumen darstellen Dabei stehen Knoten für bestimmte Taxa Kanten für eine direkte evolutionäre Verwandtschaft zwischen den beiden Knoten müssen nicht immer explizit gezeichnet werden


26 Gewurzelte und entwurzelte Bäume
27 Gewurzelte Bäume Man kann phylogenetische Bäume gewurzelt oder ungewurzelt darstellen Ein gewurzelter Baum besitzt einen Wurzelknoten, der den jüngsten gemeinsamen Vorfahr aller untersuchten Taxa darstellt Innere Knoten des Baums repräsentieren entsprechend (hypothetische) jüngste gemeinsame Vorfahren der Taxa im Zweig darunter Die Blätter des Baums entsprechen den betrachteten Taxa Der Weg von der Wurzel zu einem Blatt (Pfad) spiegelt die Evolutionsgeschichte des Taxons wieder Innerer Knoten Wurzel Ast, Zweig A B C D Blätter
28 Ungewurzelte Bäume Ungewurzelte Bäume drücken zwar die Verwandtschaft der Taxa untereinander aus, besagen aber noch nicht, wo der gemeinsame Vorfahr aller Taxa lag Gewurzelte Bäume enthalten also zusätzliche Information Einem ungewurzelten Baum entsprechen auch mithin mehrere unterschiedliche gewurzelte Bäume A C B D
29 Umwandlung
30 Wahl der Wurzel Die Umwandlung eines ungewurzelten Baums in einen gewurzelten erfordert die Auswahl einer Kante, an der die Wurzel eingefügt wird Hinzufügen eines oder mehrerer Taxa, die phylogenetisch stark unterschiedlich sind (outgroup) Vergleicht man z.b. Säugersequenzen (B1-B3), kann man entsprechende Sequenzen aus einem Fisch (A) hinzufügen Der gemeinsame Vorfahr von B1-B3 und A liegt evolutionär vor dem gemeinsamen Vorfahr von B1-B3 ) Wurzel B2 A B2 B3 B1 B3 B1 A
31 Anzahl Bäume ist exponentiell Anzahl Taxa Anzahl möglicher Bäume , , ,027, ,459, ,729, ,749,310, ,234,143, ,905,853,580,625
32 Cladogramme und Phylogramme Ein Cladogramm enthält die Information über phylogenetische Ereignisse (Verzweigungen) der dargestellten Spezies, nicht aber die Zeitinformation Phylogramme stellen an einer Achse die Zeit (oder äquivalente Größen) dar Die Lage der Knoten entspricht der (postulierten) Zeit des phylogenetischen Ereignisses B2 B2 B3 B1 A B3 B1 A T 1 T 2 T 3
33 Widersprüche in Bäumen Bestimmt man phylogenetische Bäume mit Hilfe unterschiedlicher Gene, erhält man oft unterschiedliche Bäume Gründe Unterschiedliche Mutationsraten Genduplikationen, orthologe vs. paraloge Gene Heuristiken zur Konstruktion der Bäume Horizontaler Gentransfer (nicht baumartige Evolution!) A B C D E A B C D E A B C D E 1 2 3
34 Konsensusbäume Es gibt verschiedene Arten differierende Bäume zusammenzufassen Analog zum Konsensus von Sequenzalignments, kann man auch Konsensusbäume konstruieren Oft divergieren die Bäume nur an bestimmten Stellen Diese Konsensusbäume drücken aus, welche Information in allen oder der Mehrheit der Bäume enthalten ist A B C D E A B C D E A B C D E 1 2 3
35 Strikter Konsensus Beim strikten Konsensus werden nur Verwandtschaftsbeziehungen berücksichtigt, die in allen Bäumen enthalten sind A B C D E A B C D E A B C D E A B C D E Konsensus In allen Bäumen ist der grüne Knoten Vorfahr von A, B und C B wird daher an den grünen Knoten angehängt
36 Mehrheitskonsensus Beim Mehrheitskonsensus werden alle Beziehungen übernommen, die in mehr als 50% der ursprünglichen Bäume vorkommen A B C D E A B C D E A B C D E A B C D E 67% 100% 67% Konsensus Innere Knoten, die in der Mehrzahl der Bäume auftreten werden in den Konsensusbaum übernommen

37 Reticulate Evolution Reticulate = netzartig Evolution verläuft leider nicht so geradlinig wie bisher skizziert Horizontaler Gentransfer sorgt z.b. dafür, dass eine Spezies mehrere direkte Vorfahren hat Die entstehenden Bäume sind keine Bäume mehr, sondern allgemeine Graphen, Netzwerke Entsprechend komplexer ist die phylogenetische Analyse dieser Vorgänge Mount, Bioinformatics, S. 244
38 Literatur + Links Zvelebil & Baum, Understanding Bioinformatics Mount, Bioinformatics, Kapitel 6 T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment, J. Mol. Biol. (2000), 302, T-COFFEE-Webserver
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Ringvorlesung Biologie Sommer 07 Burkhard Morgenstern Institut für Mikrobiologie und Genetik Abteilung für Bioinformatik Goldschmidtstr. 1 Online Materialien zur Ringvorlesung:
Einführung in die Bioinformatik Ringvorlesung Biologie Sommer 07 Burkhard Morgenstern Institut für Mikrobiologie und Genetik Abteilung für Bioinformatik Goldschmidtstr. 1 Online Materialien zur Ringvorlesung:
Multiple Alignments. Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann. Webseite zur Vorlesung
 Multiple Alignments Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at-
Multiple Alignments Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at-
Hidden Markov Model (HMM)
") Hidden Markov Model (HMM) Kapitel 1 Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) Jana Hertel Professur für Bioinformatik Institut für Informatik Universität Leipzig
Hidden Markov Model (HMM) Kapitel 1 Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) Jana Hertel Professur für Bioinformatik Institut für Informatik Universität Leipzig
Welche Alignmentmethoden haben Sie bisher kennengelernt?
 Welche Alignmentmethoden haben Sie bisher kennengelernt? Was heißt optimal? Optimal = die wenigsten Mutationen. Sequenzen bestehen aus Elementen (z.b. Aminosäuren oder Nukleotide). Edit Distanzen sind
Welche Alignmentmethoden haben Sie bisher kennengelernt? Was heißt optimal? Optimal = die wenigsten Mutationen. Sequenzen bestehen aus Elementen (z.b. Aminosäuren oder Nukleotide). Edit Distanzen sind
Einführung in die Angewandte Bioinformatik: Phylogenetik und Taxonomie
 Einführung in die Angewandte Bioinformatik: Phylogenetik und Taxonomie 24.06.2010 Prof. Dr. Sven Rahmann 1 Phylogenetik: Berechnung phylogenetischer Bäume Phylogenetik (phylum = Stamm): Rekonstruktion
Einführung in die Angewandte Bioinformatik: Phylogenetik und Taxonomie 24.06.2010 Prof. Dr. Sven Rahmann 1 Phylogenetik: Berechnung phylogenetischer Bäume Phylogenetik (phylum = Stamm): Rekonstruktion
TreeTOPS. Ein Phylogenetik-Icebreaker Spiel. Lehrer- Handbuch. ELLS Europäisches Lernlabor für die Lebenswissenschaften
 TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
Gleichheit, Ähnlichkeit, Homologie
 Gleichheit, Ähnlichkeit, Homologie Identität (identity) Verhältnis der Anzahl identischer Aminosäuren zur Gesamtzahl der Aminosäuren; objektiv Ähnlichkeit (similarity) Verhältnis ähnlicher Aminosäuren
Gleichheit, Ähnlichkeit, Homologie Identität (identity) Verhältnis der Anzahl identischer Aminosäuren zur Gesamtzahl der Aminosäuren; objektiv Ähnlichkeit (similarity) Verhältnis ähnlicher Aminosäuren
Bioinformatik Für Biophysiker
 Bioinformatik Für Biophysiker Sommersemester 2009 Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Schwerpunkte Algorithmen der Bioinformatik Management
Bioinformatik Für Biophysiker Sommersemester 2009 Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Schwerpunkte Algorithmen der Bioinformatik Management
Maschinelles Lernen in der Bioinformatik
 Maschinelles Lernen in der Bioinformatik Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) VL 2 HMM und (S)CFG Jana Hertel Professur für Bioinformatik Institut für Informatik
Maschinelles Lernen in der Bioinformatik Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) VL 2 HMM und (S)CFG Jana Hertel Professur für Bioinformatik Institut für Informatik
Rekonstruktion von Evolutionärer Geschichte
 Rekonstruktion von Evolutionärer Geschichte Populations- und Evolutionsbiologie 21.1.04 Florian Schiestl Phylogenetische Systematik Phylogenie: (gr. Phylum=Stamm) die Verwandtschaftsbeziehungen der Organismen,
Rekonstruktion von Evolutionärer Geschichte Populations- und Evolutionsbiologie 21.1.04 Florian Schiestl Phylogenetische Systematik Phylogenie: (gr. Phylum=Stamm) die Verwandtschaftsbeziehungen der Organismen,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de") Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Wie teilt man Lebewesen ein? 1. Versuch Aristoteles ( v.chr.)
") Taxonomie Kladistik Phylogenese Apomorphien& Co. Fachwissenschaft & Methodik bei der Ordnung und Einteilung von Lebewesen Bildquellen: http://www.ulrich-kelber.de/berlin/berlinerthemen/umwelt/biodiversitaet/index.html;
Taxonomie Kladistik Phylogenese Apomorphien& Co. Fachwissenschaft & Methodik bei der Ordnung und Einteilung von Lebewesen Bildquellen: http://www.ulrich-kelber.de/berlin/berlinerthemen/umwelt/biodiversitaet/index.html;
Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken
 Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken 14.05.2009 Prof. Dr. Sven Rahmann 1 3 Proteinsequenz-Datenbanksysteme NCBI Entrez Proteins EBI SRS Proteins UniProt (empfohlen) 2
Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken 14.05.2009 Prof. Dr. Sven Rahmann 1 3 Proteinsequenz-Datenbanksysteme NCBI Entrez Proteins EBI SRS Proteins UniProt (empfohlen) 2
Evolutionäre Bäume. Madox Sesen. 30. Juni 2014
 Evolutionäre Bäume Madox Sesen 30. Juni 2014 1 Einleitung Phylogenetische Bäume sind ein wichtiges Darstellungsmittel der Evolutionsforschung. Durch sie werden Verwandtschaftsbeziehungen zwischen Spezies
Evolutionäre Bäume Madox Sesen 30. Juni 2014 1 Einleitung Phylogenetische Bäume sind ein wichtiges Darstellungsmittel der Evolutionsforschung. Durch sie werden Verwandtschaftsbeziehungen zwischen Spezies
BCDS Seminar. Protein Tools
 BCDS Seminar Protein Tools Gliederung Nützliche Tools Three-/one-letter Amino Acids' Сodes RandSeq Random Protein Sequence Generator Protein Colourer ProtParam PeptideCutter ProtScale TMHMM Server 2.0
BCDS Seminar Protein Tools Gliederung Nützliche Tools Three-/one-letter Amino Acids' Сodes RandSeq Random Protein Sequence Generator Protein Colourer ProtParam PeptideCutter ProtScale TMHMM Server 2.0
Phylogenetik. Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann. Webseite zur Vorlesung
 Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Einführung in die Phylogenie (lat.: phylum = Stamm) Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Stammbäume Phylogenetische Bäume Evolutionsmodell
Algorithmische Bioinformatik Einführung in die Phylogenie (lat.: phylum = Stamm) Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Stammbäume Phylogenetische Bäume Evolutionsmodell
Datenstrukturen Teil 2. Bäume. Definition. Definition. Definition. Bäume sind verallgemeinerte Listen. Sie sind weiter spezielle Graphen
 Bäume sind verallgemeinerte Listen Datenstrukturen Teil 2 Bäume Jeder Knoten kann mehrere Nachfolger haben Sie sind weiter spezielle Graphen Graphen bestehen aus Knoten und Kanten Kanten können gerichtet
Bäume sind verallgemeinerte Listen Datenstrukturen Teil 2 Bäume Jeder Knoten kann mehrere Nachfolger haben Sie sind weiter spezielle Graphen Graphen bestehen aus Knoten und Kanten Kanten können gerichtet
Zentrum für Bioinformatik. Übung 4: Revision. Beispielfragen zur Klausur im Modul Angewandte Bioinformatik (erste Semesterhälfte)
") Andrew Torda Björn Hansen Iryna Bondarenko Zentrum für Bioinformatik Übung zur Vorlesung Angewandte Bioinformatik Sommersemester 2014 20./23.06.2014 Übung 4: Revision Beispielfragen zur Klausur im Modul
Andrew Torda Björn Hansen Iryna Bondarenko Zentrum für Bioinformatik Übung zur Vorlesung Angewandte Bioinformatik Sommersemester 2014 20./23.06.2014 Übung 4: Revision Beispielfragen zur Klausur im Modul
Diskrete Strukturen Kapitel 4: Graphentheorie (Bäume)
") WS 2016/17 Diskrete Strukturen Kapitel 4: Graphentheorie (Bäume) Hans-Joachim Bungartz Lehrstuhl für wissenschaftliches Rechnen Fakultät für Informatik Technische Universität München http://www5.in.tum.de/wiki/index.php/diskrete_strukturen_-_winter_16
WS 2016/17 Diskrete Strukturen Kapitel 4: Graphentheorie (Bäume) Hans-Joachim Bungartz Lehrstuhl für wissenschaftliches Rechnen Fakultät für Informatik Technische Universität München http://www5.in.tum.de/wiki/index.php/diskrete_strukturen_-_winter_16
Evolutionary Trees: Distance Based
 Evolutionary Trees: Distance Based 1 Buftea Alexandru Laut der Evolutionstheorie findet in allen Organismen eine langsame Änderung statt (Evolution). Ein evolutionärer Baum, auch phylogenetischer Baum
Evolutionary Trees: Distance Based 1 Buftea Alexandru Laut der Evolutionstheorie findet in allen Organismen eine langsame Änderung statt (Evolution). Ein evolutionärer Baum, auch phylogenetischer Baum
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
MOL.504 Analyse von DNA- und Proteinsequenzen. Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
WS 2013/14. Diskrete Strukturen
 WS 2013/14 Diskrete Strukturen Prof. Dr. J. Esparza Lehrstuhl für Grundlagen der Softwarezuverlässigkeit und theoretische Informatik Fakultät für Informatik Technische Universität München http://www7.in.tum.de/um/courses/ds/ws1314
WS 2013/14 Diskrete Strukturen Prof. Dr. J. Esparza Lehrstuhl für Grundlagen der Softwarezuverlässigkeit und theoretische Informatik Fakultät für Informatik Technische Universität München http://www7.in.tum.de/um/courses/ds/ws1314
Bioinformatik Für Biophysiker
 Bioinformatik Für Biophysiker Wintersemester 2005 / 2006 Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Lehrstuhl seit 10/2002 Schwerpunkte Algorithmen der Bioinformatik
Bioinformatik Für Biophysiker Wintersemester 2005 / 2006 Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Lehrstuhl seit 10/2002 Schwerpunkte Algorithmen der Bioinformatik
Matrizen. Nicht nur eine Matrix, sondern viele 0,5 0,2 0,3 A 0,2 0,7 0,1
 Nicht nur eine Matrix, sondern viele Matrizen 0,5 0,2 0,3 A 0,2 0,7 0,1 015 0,15 0,75 075 0,1 01 aber keine Matrize und auch keine Matratzen 1 Wie beschreibt man Prozesse? Makov-Modell Modell Markov- Prozess
Nicht nur eine Matrix, sondern viele Matrizen 0,5 0,2 0,3 A 0,2 0,7 0,1 015 0,15 0,75 075 0,1 01 aber keine Matrize und auch keine Matratzen 1 Wie beschreibt man Prozesse? Makov-Modell Modell Markov- Prozess
Phylogenetische Analyse
 Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Studiengang Informatik der FH Gießen-Friedberg. Sequenz-Alignment. Jan Schäfer. WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel
 Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
Bioinformatik. Lokale Alignierung Gapkosten. Silke Trißl / Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
9 Minimum Spanning Trees
 Im Folgenden wollen wir uns genauer mit dem Minimum Spanning Tree -Problem auseinandersetzen. 9.1 MST-Problem Gegeben ein ungerichteter Graph G = (V,E) und eine Gewichtsfunktion w w : E R Man berechne
Im Folgenden wollen wir uns genauer mit dem Minimum Spanning Tree -Problem auseinandersetzen. 9.1 MST-Problem Gegeben ein ungerichteter Graph G = (V,E) und eine Gewichtsfunktion w w : E R Man berechne
Darwins Erben - Phylogenie und Bäume
 Vorlesung Einführung in die Bioinforma4k SoSe2011 Darwins Erben - Phylogenie und Bäume Prof. Daniel Huson ZBIT Center for Bioinformatics Center for Bioinformatics Charles Darwin und Bäume Darwin's Notizbuch
Vorlesung Einführung in die Bioinforma4k SoSe2011 Darwins Erben - Phylogenie und Bäume Prof. Daniel Huson ZBIT Center for Bioinformatics Center for Bioinformatics Charles Darwin und Bäume Darwin's Notizbuch
Bayesianische Netzwerke - Lernen und Inferenz
 Bayesianische Netzwerke - Lernen und Inferenz Manuela Hummel 9. Mai 2003 Gliederung 1. Allgemeines 2. Bayesianische Netzwerke zur Auswertung von Genexpressionsdaten 3. Automatische Modellselektion 4. Beispiel
Bayesianische Netzwerke - Lernen und Inferenz Manuela Hummel 9. Mai 2003 Gliederung 1. Allgemeines 2. Bayesianische Netzwerke zur Auswertung von Genexpressionsdaten 3. Automatische Modellselektion 4. Beispiel
Tutoraufgabe 1 (Suchen in Graphen):
:") Prof. aa Dr. E. Ábrahám Datenstrukturen und Algorithmen SS14 F. Corzilius, S. Schupp, T. Ströder Tutoraufgabe 1 (Suchen in Graphen): a) Geben Sie die Reihenfolge an, in der die Knoten besucht werden, wenn
Prof. aa Dr. E. Ábrahám Datenstrukturen und Algorithmen SS14 F. Corzilius, S. Schupp, T. Ströder Tutoraufgabe 1 (Suchen in Graphen): a) Geben Sie die Reihenfolge an, in der die Knoten besucht werden, wenn
Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments
 Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments Sven Rahmann Genominformatik Universitätsklinikum Essen Universität Duisburg-Essen Universitätsallianz Ruhr Einführung Bisher: Berechnung
Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments Sven Rahmann Genominformatik Universitätsklinikum Essen Universität Duisburg-Essen Universitätsallianz Ruhr Einführung Bisher: Berechnung
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Center-Star Score Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Multiples Sequenzalignment Sum-Of-Pair
Algorithmische Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Center-Star Score Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Multiples Sequenzalignment Sum-Of-Pair
Logik für Informatiker
 Vorlesung Logik für Informatiker 13. Prädikatenlogik Der Satz von Herbrand Bernhard Beckert Universität Koblenz-Landau Sommersemester 2006 Logik für Informatiker, SS 06 p.1 Semantische Bäume Eine klassische
Vorlesung Logik für Informatiker 13. Prädikatenlogik Der Satz von Herbrand Bernhard Beckert Universität Koblenz-Landau Sommersemester 2006 Logik für Informatiker, SS 06 p.1 Semantische Bäume Eine klassische
Bioinformatik II: Phylogenetik
 Bioinformatik II: Phylogenetik phylogenetisch Phylai: griechische Klans phylum: der Stamm phylogenetisch: die Stammesgeschichte von Lebewesen betreffend Hierarchien der Klassifikation: Domäne: Eukaryonten
Bioinformatik II: Phylogenetik phylogenetisch Phylai: griechische Klans phylum: der Stamm phylogenetisch: die Stammesgeschichte von Lebewesen betreffend Hierarchien der Klassifikation: Domäne: Eukaryonten
Alle bislang betrachteten Sortieralgorithmen hatten (worst-case) Laufzeit Ω(nlog(n)).
 Laufzeit Ω(nlog(n)).") 8. Untere Schranken für Sortieren Alle bislang betrachteten Sortieralgorithmen hatten (worst-case) Laufzeit Ω(nlog(n)). Werden nun gemeinsame Eigenschaften dieser Algorithmen untersuchen. Fassen gemeinsame
8. Untere Schranken für Sortieren Alle bislang betrachteten Sortieralgorithmen hatten (worst-case) Laufzeit Ω(nlog(n)). Werden nun gemeinsame Eigenschaften dieser Algorithmen untersuchen. Fassen gemeinsame
Graphentheorie Graphentheorie. Grundlagen Bäume Eigenschaften von Graphen Graphen-Algorithmen Matchings und Netzwerke
 Graphen Graphentheorie Graphentheorie Grundlagen Bäume Eigenschaften von Graphen Graphen-Algorithmen Matchings und Netzwerke 2 Was ist ein Graph? Ein Graph ist in der Graphentheorie eine abstrakte Struktur,
Graphen Graphentheorie Graphentheorie Grundlagen Bäume Eigenschaften von Graphen Graphen-Algorithmen Matchings und Netzwerke 2 Was ist ein Graph? Ein Graph ist in der Graphentheorie eine abstrakte Struktur,
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 06. Paarweises Alignment Teil II Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 06. Paarweises Alignment Teil II Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Informationsvisualisierung
 Informationsvisualisierung Thema: 7. Visualisierung Biologischer Daten Dozent: Dr. Dirk Zeckzer zeckzer@informatik.uni-leipzig.de Sprechstunde: nach Vereinbarung Umfang: 2 Prüfungsfach: Modul Fortgeschrittene
Informationsvisualisierung Thema: 7. Visualisierung Biologischer Daten Dozent: Dr. Dirk Zeckzer zeckzer@informatik.uni-leipzig.de Sprechstunde: nach Vereinbarung Umfang: 2 Prüfungsfach: Modul Fortgeschrittene
Algorithmen und Datenstrukturen II
 Algorithmen und Datenstrukturen II Algorithmen zur Textverarbeitung III: D. Rösner Institut für Wissens- und Sprachverarbeitung Fakultät für Informatik Otto-von-Guericke Universität Magdeburg Sommer 2009,
Algorithmen und Datenstrukturen II Algorithmen zur Textverarbeitung III: D. Rösner Institut für Wissens- und Sprachverarbeitung Fakultät für Informatik Otto-von-Guericke Universität Magdeburg Sommer 2009,
1 Schulinterner Kernlehrplan Biologie Q2 Evolution
 1 Schulinterner Kernlehrplan Biologie Q2 Evolution 1 Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen Evolution Evolutionstheorien LK Evolutionstheorie Biodiversität und Systematik Entwicklung
1 Schulinterner Kernlehrplan Biologie Q2 Evolution 1 Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen Evolution Evolutionstheorien LK Evolutionstheorie Biodiversität und Systematik Entwicklung
Softwarewerkzeuge der Bioinformatik
 Bioinformatik Wintersemester 2006/2007 Tutorial 2: paarweise Sequenzaligments BLAST Tutorial 2: BLAST 1/22 Alignment Ausrichten zweier oder mehrerer Sequenzen, um: ihre Ähnlichkeit quantitativ zu erfassen
Bioinformatik Wintersemester 2006/2007 Tutorial 2: paarweise Sequenzaligments BLAST Tutorial 2: BLAST 1/22 Alignment Ausrichten zweier oder mehrerer Sequenzen, um: ihre Ähnlichkeit quantitativ zu erfassen
InterPro & SP-ML. Syntax und Verwendung der Beschreibungssprache XML Ausarbeitung im Seminar XML in der Bioinformatik.
 InterPro & SP-ML Syntax und Verwendung der Beschreibungssprache XML Ausarbeitung im Seminar XML in der Bioinformatik Stefan Albaum 18. Dezember 2002 Inhaltsverzeichnis 1 SPTr-XML 2 1.1 SWISS-PROT...........................
InterPro & SP-ML Syntax und Verwendung der Beschreibungssprache XML Ausarbeitung im Seminar XML in der Bioinformatik Stefan Albaum 18. Dezember 2002 Inhaltsverzeichnis 1 SPTr-XML 2 1.1 SWISS-PROT...........................
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 5. Paarweises Alignment Teil I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 5. Paarweises Alignment Teil I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Algorithmische Anwendungen WS 2005/2006
 Algorithmische Anwendungen WS 2005/2006 Sequenzalignment Gruppe F_lila_Ala0506 Allal Kharaz Yassine ELassad Inhaltsverzeichnis 1 Problemstellungen...................................... 3 1.1 Rechtschreibkorrektur...............................
Algorithmische Anwendungen WS 2005/2006 Sequenzalignment Gruppe F_lila_Ala0506 Allal Kharaz Yassine ELassad Inhaltsverzeichnis 1 Problemstellungen...................................... 3 1.1 Rechtschreibkorrektur...............................
Primärstruktur. Wintersemester 2011/12. Peter Güntert
 Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2012 5. Biologische Netzwerke - Gut vernetzt hält besser Überblick
Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2012 5. Biologische Netzwerke - Gut vernetzt hält besser Überblick
Fortgeschrittene Netzwerk- und Graph-Algorithmen
 Fortgeschrittene Netzwerk- und Graph-Algorithmen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Wintersemester
Fortgeschrittene Netzwerk- und Graph-Algorithmen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Wintersemester
Was ist eine Art? Molekulare Methoden zur Erfassung genetischer Diversität. Nadine Bernhardt Experimentelle Taxonomie
 Was ist eine Art? Molekulare Methoden zur Erfassung genetischer Diversität Nadine Bernhardt Experimentelle Taxonomie 10.10.2016 IPK Gatersleben Sam Rey/IPK IPK Gatersleben: Genbank Erhaltung und Nutzbarmachung
Was ist eine Art? Molekulare Methoden zur Erfassung genetischer Diversität Nadine Bernhardt Experimentelle Taxonomie 10.10.2016 IPK Gatersleben Sam Rey/IPK IPK Gatersleben: Genbank Erhaltung und Nutzbarmachung
Binary Decision Diagrams (Einführung)
") Binary Decision Diagrams (Einführung) Binary Decision Diagrams (BDDs) sind bestimmte Graphen, die als Datenstruktur für die kompakte Darstellung von booleschen Funktionen benutzt werden. BDDs wurden von
Binary Decision Diagrams (Einführung) Binary Decision Diagrams (BDDs) sind bestimmte Graphen, die als Datenstruktur für die kompakte Darstellung von booleschen Funktionen benutzt werden. BDDs wurden von
Attached! Proseminar Netzwerkanalyse SS 2004 Thema: Biologie
 Rheinisch-Westfälischen Technischen Hochschule Aachen Lehr- und Forschungsgebiet Theoretische Informatik Prof. Rossmanith Attached! Proseminar Netzwerkanalyse SS 2004 Thema: Biologie Deniz Özmen Emmanuel
Rheinisch-Westfälischen Technischen Hochschule Aachen Lehr- und Forschungsgebiet Theoretische Informatik Prof. Rossmanith Attached! Proseminar Netzwerkanalyse SS 2004 Thema: Biologie Deniz Özmen Emmanuel
Statistische Verfahren:
 Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Molekulare Bioinformatik
 Molekulare Bioinformatik Wintersemester 203/204 Prof. Thomas Martinetz Institut für Neuro- und Bioinformatik Universität zu Luebeck 07.0.204 Molekulare Bioinformatik - Vorlesung 0 Wiederhohlung Die Entropie
Molekulare Bioinformatik Wintersemester 203/204 Prof. Thomas Martinetz Institut für Neuro- und Bioinformatik Universität zu Luebeck 07.0.204 Molekulare Bioinformatik - Vorlesung 0 Wiederhohlung Die Entropie
3.1.1 Die Variante T1 und ein Entscheidungsverfahren für die Aussagenlogik
 Deduktionssysteme der Aussagenlogik, Kap. 3: Tableaukalküle 38 3 Tableaukalküle 3.1 Klassische Aussagenlogik 3.1.1 Die Variante T1 und ein Entscheidungsverfahren für die Aussagenlogik Ein zweites Entscheidungsverfahren
Deduktionssysteme der Aussagenlogik, Kap. 3: Tableaukalküle 38 3 Tableaukalküle 3.1 Klassische Aussagenlogik 3.1.1 Die Variante T1 und ein Entscheidungsverfahren für die Aussagenlogik Ein zweites Entscheidungsverfahren
Kapitel 4: Netzplantechnik Gliederung der Vorlesung
 Gliederung der Vorlesung 1. Grundbegriffe 2. Elementare Graphalgorithmen und Anwendungen 3. Kürzeste Wege 4. Netzplantechnik 5. Minimal spannende Bäume 6. Traveling Salesman Problem 7. Flüsse in Netzwerken
Gliederung der Vorlesung 1. Grundbegriffe 2. Elementare Graphalgorithmen und Anwendungen 3. Kürzeste Wege 4. Netzplantechnik 5. Minimal spannende Bäume 6. Traveling Salesman Problem 7. Flüsse in Netzwerken
Bioinformatik. Substitutionsmatrizen BLAST. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Hidden-Markov-Modelle
 Proseminar: Machine-Learning Hidden-Markov-Modelle Benjamin Martin Zusammenfassung 1953 stellten Watson und Crick ihr DNA-Modell vor. Damit öffnete sich für Genforscher ein riesiges Gebiet, das bisher
Proseminar: Machine-Learning Hidden-Markov-Modelle Benjamin Martin Zusammenfassung 1953 stellten Watson und Crick ihr DNA-Modell vor. Damit öffnete sich für Genforscher ein riesiges Gebiet, das bisher
TECHNISCHE UNIVERSITÄT MÜNCHEN FAKULTÄT FÜR INFORMATIK
 TECHNISCHE UNIVERSITÄT MÜNCHEN FAKULTÄT FÜR INFORMATIK Lehrstuhl für Sprachen und Beschreibungsstrukturen SS 2009 Grundlagen: Algorithmen und Datenstrukturen Übungsblatt 11 Prof. Dr. Helmut Seidl, S. Pott,
TECHNISCHE UNIVERSITÄT MÜNCHEN FAKULTÄT FÜR INFORMATIK Lehrstuhl für Sprachen und Beschreibungsstrukturen SS 2009 Grundlagen: Algorithmen und Datenstrukturen Übungsblatt 11 Prof. Dr. Helmut Seidl, S. Pott,
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2011 8. Biologische Netzwerke - Gut vernetzt hält besser Überblick
Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2011 8. Biologische Netzwerke - Gut vernetzt hält besser Überblick
Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
 Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof Daniel Stöckel, M. Sc. Patrick Trampert, M. Sc. Lara Schneider, M. Sc. I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof Daniel Stöckel, M. Sc. Patrick Trampert, M. Sc. Lara Schneider, M. Sc. I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
FOR MUW SSM3 (2008) STUDENTS EDUCATIONAL PURPOSE ONLY
 STUDENTS EDUCATIONAL PURPOSE ONLY") Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
MBI: Sequenzvergleich ohne Alignment
 MBI: Sequenzvergleich ohne Alignment Bernhard Haubold 12. November 2013 Wiederholung Exaktes & inexaktes Matching Das exakte Matching Problem Naive Lösung Präprozessierung Muster(Pattern): Z-Algorithmus,
MBI: Sequenzvergleich ohne Alignment Bernhard Haubold 12. November 2013 Wiederholung Exaktes & inexaktes Matching Das exakte Matching Problem Naive Lösung Präprozessierung Muster(Pattern): Z-Algorithmus,
Bioinformatik. BLAST Basic Local Alignment Search Tool. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik BLAST Basic Local Alignment Search Tool Ulf Leser Wissensmanagement in der Bioinformatik Heuristische Alignierung Annotation neuer Sequenzen basiert auf Suche nach homologen Sequenzen in
Bioinformatik BLAST Basic Local Alignment Search Tool Ulf Leser Wissensmanagement in der Bioinformatik Heuristische Alignierung Annotation neuer Sequenzen basiert auf Suche nach homologen Sequenzen in
Sequence Assembly. Nicola Palandt
 Sequence Assembly Nicola Palandt 1 Einleitung Das Genom eines Lebewesens ist der Träger aller Informationen, die eine Zelle weitergeben kann. Es besteht aus Sequenzen, die mehrere Milliarden Basen lang
Sequence Assembly Nicola Palandt 1 Einleitung Das Genom eines Lebewesens ist der Träger aller Informationen, die eine Zelle weitergeben kann. Es besteht aus Sequenzen, die mehrere Milliarden Basen lang
Graphentheorie. Eulersche Graphen. Eulersche Graphen. Eulersche Graphen. Rainer Schrader. 14. November Gliederung.
 Graphentheorie Rainer Schrader Zentrum für Angewandte Informatik Köln 14. November 2007 1 / 22 2 / 22 Gliederung eulersche und semi-eulersche Graphen Charakterisierung eulerscher Graphen Berechnung eines
Graphentheorie Rainer Schrader Zentrum für Angewandte Informatik Köln 14. November 2007 1 / 22 2 / 22 Gliederung eulersche und semi-eulersche Graphen Charakterisierung eulerscher Graphen Berechnung eines
Algorithmen für paarweise Sequenz-Alignments. Katharina Hembach
 Proseminar Bioinformatik WS 2010/11 Algorithmen für paarweise Sequenz-Alignments Katharina Hembach 06.12.2010 1 Einleitung Paarweise Sequenz-Alignments spielen in der Bioinformatik eine wichtige Rolle.
Proseminar Bioinformatik WS 2010/11 Algorithmen für paarweise Sequenz-Alignments Katharina Hembach 06.12.2010 1 Einleitung Paarweise Sequenz-Alignments spielen in der Bioinformatik eine wichtige Rolle.
Molekulare Evolution
 Molekulare Evolution 1. Grundprinzip: Die Evolution arbeitet aus gegebenen Materialien neue Strukturen (Gene, Organe, usw.) enstehen nur aus schon existierenden Technische vs. Biologische 1. Technische
Molekulare Evolution 1. Grundprinzip: Die Evolution arbeitet aus gegebenen Materialien neue Strukturen (Gene, Organe, usw.) enstehen nur aus schon existierenden Technische vs. Biologische 1. Technische
8.4 Suffixbäume. Anwendungen: Information Retrieval, Bioinformatik (Suche in Sequenzen) Veranschaulichung: DNA-Sequenzen
 Veranschaulichung: DNA-Sequenzen") 8.4 Suffixbäume Ziel: Datenstruktur, die effiziente Operationen auf (langen) Zeichenketten unterstützt: - Suche Teilzeichenkette (Substring) - Präfix - längste sich wiederholende Zeichenkette -... Anwendungen:
8.4 Suffixbäume Ziel: Datenstruktur, die effiziente Operationen auf (langen) Zeichenketten unterstützt: - Suche Teilzeichenkette (Substring) - Präfix - längste sich wiederholende Zeichenkette -... Anwendungen:
DynaTraffic Modelle und mathematische Prognosen. Simulation der Verteilung des Verkehrs mit Hilfe von Markov-Ketten
 DynaTraffic Modelle und mathematische Prognosen Simulation der Verteilung des Verkehrs mit Hilfe von Markov-Ketten Worum geht es? Modelle von Verkehrssituationen Graphen: Kanten, Knoten Matrixdarstellung
DynaTraffic Modelle und mathematische Prognosen Simulation der Verteilung des Verkehrs mit Hilfe von Markov-Ketten Worum geht es? Modelle von Verkehrssituationen Graphen: Kanten, Knoten Matrixdarstellung
Grundlagen: Algorithmen und Datenstrukturen
 Grundlagen: Algorithmen und Datenstrukturen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2010
Grundlagen: Algorithmen und Datenstrukturen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2010
Methoden der KI in der Biomedizin Bayes Netze
 Methoden der KI in der Biomedizin Bayes Netze Karl D. Fritscher Bayes Netze Intuitiv: Graphische Repräsentation von Einfluss Mathematisch: Graphische Repräsentation von bedingter Unabhängigkeit Bayes Netze
Methoden der KI in der Biomedizin Bayes Netze Karl D. Fritscher Bayes Netze Intuitiv: Graphische Repräsentation von Einfluss Mathematisch: Graphische Repräsentation von bedingter Unabhängigkeit Bayes Netze
Allgemeine Aufgabenstellung. Ziele
 Allgemeine Aufgabenstellung Sie (s)wollen die Phylogenie der Vertebraten mit Hilfe molekulare Daten ergründen. Insbesondere interessiert Sie die Verwandtschaft der Primaten; aber auch tiefere Verzweigungen
Allgemeine Aufgabenstellung Sie (s)wollen die Phylogenie der Vertebraten mit Hilfe molekulare Daten ergründen. Insbesondere interessiert Sie die Verwandtschaft der Primaten; aber auch tiefere Verzweigungen
Wiederholung. Bäume sind zyklenfrei. Rekursive Definition: Baum = Wurzelknoten + disjunkte Menge von Kindbäumen.
 Wiederholung Baum: Gerichteter Graph, der die folgenden drei Bedingungen erfüllt: Es gibt einen Knoten, der nicht Endknoten einer Kante ist. (Dieser Knoten heißt Wurzel des Baums.) Jeder andere Knoten
Wiederholung Baum: Gerichteter Graph, der die folgenden drei Bedingungen erfüllt: Es gibt einen Knoten, der nicht Endknoten einer Kante ist. (Dieser Knoten heißt Wurzel des Baums.) Jeder andere Knoten
Kapitel 7: Flüsse in Netzwerken und Anwendungen Gliederung der Vorlesung
 Gliederung der Vorlesung. Fallstudie Bipartite Graphen. Grundbegriffe 3. Elementare Graphalgorithmen und Anwendungen 4. Minimal spannende Bäume 5. Kürzeste Pfade 6. Traveling Salesman Problem 7. Flüsse
Gliederung der Vorlesung. Fallstudie Bipartite Graphen. Grundbegriffe 3. Elementare Graphalgorithmen und Anwendungen 4. Minimal spannende Bäume 5. Kürzeste Pfade 6. Traveling Salesman Problem 7. Flüsse
Vorlesung Informatik 2 Algorithmen und Datenstrukturen
 Vorlesung Informatik 2 Algorithmen und Datenstrukturen (18 Bäume: Grundlagen und natürliche Suchbäume) Prof. Dr. Susanne Albers Bäume (1) Bäume sind verallgemeinerte Listen (jedes Knoten-Element kann mehr
Vorlesung Informatik 2 Algorithmen und Datenstrukturen (18 Bäume: Grundlagen und natürliche Suchbäume) Prof. Dr. Susanne Albers Bäume (1) Bäume sind verallgemeinerte Listen (jedes Knoten-Element kann mehr
V3 - Multiples Sequenz Alignment und Phylogenie
 V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 1 Leitfragen für V3 Frage1: Können wir aus dem Vergleich
V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 1 Leitfragen für V3 Frage1: Können wir aus dem Vergleich
Formale Methoden 1. Gerhard Jäger 12. Dezember Uni Bielefeld, WS 2007/2008 1/22
 1/22 Formale Methoden 1 Gerhard Jäger Gerhard.Jaeger@uni-bielefeld.de Uni Bielefeld, WS 2007/2008 12. Dezember 2007 2/22 Bäume Baumdiagramme Ein Baumdiagramm eines Satzes stellt drei Arten von Information
1/22 Formale Methoden 1 Gerhard Jäger Gerhard.Jaeger@uni-bielefeld.de Uni Bielefeld, WS 2007/2008 12. Dezember 2007 2/22 Bäume Baumdiagramme Ein Baumdiagramm eines Satzes stellt drei Arten von Information
Hidden Markov Models
 Hidden Markov Models Kursfolien Karin Haenelt 09.05002 1 Letzte Änderung 18.07002 Hidden Markov Models Besondere Form eines probabilistischen endlichen Automaten Weit verbreitet in der statistischen Sprachverarbeitung
Hidden Markov Models Kursfolien Karin Haenelt 09.05002 1 Letzte Änderung 18.07002 Hidden Markov Models Besondere Form eines probabilistischen endlichen Automaten Weit verbreitet in der statistischen Sprachverarbeitung
Dynamisches Huffman-Verfahren
 Dynamisches Huffman-Verfahren - Adaptive Huffman Coding - von Michael Brückner 1. Einleitung 2. Der Huffman-Algorithmus 3. Übergang zu einem dynamischen Verfahren 4. Der FGK-Algorithmus 5. Überblick über
Dynamisches Huffman-Verfahren - Adaptive Huffman Coding - von Michael Brückner 1. Einleitung 2. Der Huffman-Algorithmus 3. Übergang zu einem dynamischen Verfahren 4. Der FGK-Algorithmus 5. Überblick über
TU München. Hauptseminar: WS 2002 / Einführung in Suffix - Bäume
 TU München Hauptseminar: WS 2002 / 2003 Einführung in Suffix - Bäume Bearbeiterin: Shasha Meng Betreuerin: Barbara König Inhalt 1. Einleitung 1.1 Motivation 1.2 Eine kurze Geschichte 2. Tries 2.1 Basisdefinition
TU München Hauptseminar: WS 2002 / 2003 Einführung in Suffix - Bäume Bearbeiterin: Shasha Meng Betreuerin: Barbara König Inhalt 1. Einleitung 1.1 Motivation 1.2 Eine kurze Geschichte 2. Tries 2.1 Basisdefinition
Featurebasierte 3D Modellierung
 1 Featurebasierte 3D Modellierung Moderne 3D arbeiten häufig mit einer Feature Modellierung. Hierbei gibt es eine Reihe von vordefinierten Konstruktionen, die der Reihe nach angewandt werden. Diese Basis
1 Featurebasierte 3D Modellierung Moderne 3D arbeiten häufig mit einer Feature Modellierung. Hierbei gibt es eine Reihe von vordefinierten Konstruktionen, die der Reihe nach angewandt werden. Diese Basis
Sachrechnen/Größen WS 14/15-
 Kapitel Daten & Wahrscheinlichkeit 3.1 Kombinatorische Grundlagen 3.2 Kombinatorik & Wahrscheinlichkeit in der Grundschule 3.3 Daten Darstellen 3.1 Kombinatorische Grundlagen Verschiedene Bereiche der
Kapitel Daten & Wahrscheinlichkeit 3.1 Kombinatorische Grundlagen 3.2 Kombinatorik & Wahrscheinlichkeit in der Grundschule 3.3 Daten Darstellen 3.1 Kombinatorische Grundlagen Verschiedene Bereiche der
Digraphen, DAGs und Wurzelbäume
 Digraphen (gerichtete Graphen) Slide 1 Digraphen, DAGs und Wurzelbäume Digraphen (gerichtete Graphen) Slide 2 Eingangs- und Ausgangsgrad Bei einer gerichteten Kante e = (u,v) E heißt u Startknoten von
Digraphen (gerichtete Graphen) Slide 1 Digraphen, DAGs und Wurzelbäume Digraphen (gerichtete Graphen) Slide 2 Eingangs- und Ausgangsgrad Bei einer gerichteten Kante e = (u,v) E heißt u Startknoten von
Kapitel 5: Minimale spannende Bäume Gliederung der Vorlesung
 Gliederung der Vorlesung 1. Grundbegriffe 2. Elementare Graphalgorithmen und Anwendungen 3. Kürzeste Wege. Minimale spannende Bäume. Färbungen und Cliquen. Traveling Salesman Problem. Flüsse in Netzwerken
Gliederung der Vorlesung 1. Grundbegriffe 2. Elementare Graphalgorithmen und Anwendungen 3. Kürzeste Wege. Minimale spannende Bäume. Färbungen und Cliquen. Traveling Salesman Problem. Flüsse in Netzwerken
Theoretische Informatik. nichtdeterministische Turingmaschinen NDTM. Turingmaschinen. Rainer Schrader. 29. April 2009
 Theoretische Informatik Rainer Schrader nichtdeterministische Turingmaschinen Zentrum für Angewandte Informatik Köln 29. April 2009 1 / 33 2 / 33 Turingmaschinen das Konzept des Nichtdeterminismus nahm
Theoretische Informatik Rainer Schrader nichtdeterministische Turingmaschinen Zentrum für Angewandte Informatik Köln 29. April 2009 1 / 33 2 / 33 Turingmaschinen das Konzept des Nichtdeterminismus nahm
Theoretische Informatik 1
 Theoretische Informatik 1 Approximierbarkeit David Kappel Institut für Grundlagen der Informationsverarbeitung Technische Universität Graz 10.06.2016 Übersicht Das Problem des Handelsreisenden TSP EUCLIDEAN-TSP
Theoretische Informatik 1 Approximierbarkeit David Kappel Institut für Grundlagen der Informationsverarbeitung Technische Universität Graz 10.06.2016 Übersicht Das Problem des Handelsreisenden TSP EUCLIDEAN-TSP
Bioinformatik. Distanzbasierte phylogenetische Algorithmen. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Distanzbasierte phylogenetische Algorithmen Ulf Leser Wissensmanagement in der Bioinformatik Phylogenetische Bäume Stammbaum (Phylogenetic Tree) Ulf Leser: Algorithmische Bioinformatik, Wintersemester
Bioinformatik Distanzbasierte phylogenetische Algorithmen Ulf Leser Wissensmanagement in der Bioinformatik Phylogenetische Bäume Stammbaum (Phylogenetic Tree) Ulf Leser: Algorithmische Bioinformatik, Wintersemester
Clausthal C G C C G C. Informatik II Bäume. G. Zachmann Clausthal University, Germany Beispiele. Stammbaum.
 lausthal Informatik II Bäume. Zachmann lausthal University, ermany zach@in.tu-clausthal.de Beispiele Stammbaum. Zachmann Informatik - SS 0 Bäume Stammbaum Parse tree, Rekursionsbaum Unix file hierarchy
lausthal Informatik II Bäume. Zachmann lausthal University, ermany zach@in.tu-clausthal.de Beispiele Stammbaum. Zachmann Informatik - SS 0 Bäume Stammbaum Parse tree, Rekursionsbaum Unix file hierarchy
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2014 8. Biologische Netzwerke - Gut vernetzt hält besser Überblick
Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2014 8. Biologische Netzwerke - Gut vernetzt hält besser Überblick
WS 2009/10. Diskrete Strukturen
 WS 2009/10 Diskrete Strukturen Prof. Dr. J. Esparza Lehrstuhl für Grundlagen der Softwarezuverlässigkeit und theoretische Informatik Fakultät für Informatik Technische Universität München http://www7.in.tum.de/um/courses/ds/ws0910
WS 2009/10 Diskrete Strukturen Prof. Dr. J. Esparza Lehrstuhl für Grundlagen der Softwarezuverlässigkeit und theoretische Informatik Fakultät für Informatik Technische Universität München http://www7.in.tum.de/um/courses/ds/ws0910
V3 - Multiples Sequenz Alignment und Phylogenie
 V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 3. Vorlesung SS 2011 Softwarewerkzeuge der Bioinformatik
V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 3. Vorlesung SS 2011 Softwarewerkzeuge der Bioinformatik
Stammesgeschichte des Menschen
 Winfried Henke Hartmut Rothe Stammesgeschichte des Menschen Eine Einführung Mit 87 Abbildungen Springer 1 Von der Schöpfungs- zur Stammesgeschichte. 2 Allgemeine Grundlagen der Verwandtschaftsanalyse 7
Winfried Henke Hartmut Rothe Stammesgeschichte des Menschen Eine Einführung Mit 87 Abbildungen Springer 1 Von der Schöpfungs- zur Stammesgeschichte. 2 Allgemeine Grundlagen der Verwandtschaftsanalyse 7
Homologie und Sequenzähnlichkeit. Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Mündliche Prüfung Kurs Grundlegende Algorithmen der Bioinformatik 30 Minuten, , Prof. Merkl
 Erinnerungsprotokoll Mündliche Prüfung Kurs 01738 Grundlegende Algorithmen der Bioinformatik 30 Minuten, 13.05.2015, Prof. Merkl - Zuerst zu Taxonomie. Was ist das und warum macht man das? Finden einer
Erinnerungsprotokoll Mündliche Prüfung Kurs 01738 Grundlegende Algorithmen der Bioinformatik 30 Minuten, 13.05.2015, Prof. Merkl - Zuerst zu Taxonomie. Was ist das und warum macht man das? Finden einer
Kapitel 2: Sortier- und Selektionsverfahren Gliederung
 Gliederung 1. Laufzeit von Algorithmen 2. Sortier- und Selektionsverfahren 3. Paradigmen des Algorithmenentwurfs 4. Ausgewählte Datenstrukturen 5. Algorithmische Geometrie 6. Randomisierte Algorithmen
Gliederung 1. Laufzeit von Algorithmen 2. Sortier- und Selektionsverfahren 3. Paradigmen des Algorithmenentwurfs 4. Ausgewählte Datenstrukturen 5. Algorithmische Geometrie 6. Randomisierte Algorithmen
Algorithmen und Datenstrukturen 1
 Algorithmen und Datenstrukturen 1 6. Vorlesung Martin Middendorf / Universität Leipzig Institut für Informatik middendorf@informatik.uni-leipzig.de studla@bioinf.uni-leipzig.de Merge-Sort Anwendbar für
Algorithmen und Datenstrukturen 1 6. Vorlesung Martin Middendorf / Universität Leipzig Institut für Informatik middendorf@informatik.uni-leipzig.de studla@bioinf.uni-leipzig.de Merge-Sort Anwendbar für
Evolutionsbiologie 1 Molekulare Evolution Sebastian Höhna
 Evolutionsbiologie 1 Molekulare Evolution Sebastian Höhna Division of Evolutionary Biology Ludwig-Maximilians Universität, München Überblick: Molekulare Analysen 1) Generierung von Sequenzdaten: a) Sequenzierung
Evolutionsbiologie 1 Molekulare Evolution Sebastian Höhna Division of Evolutionary Biology Ludwig-Maximilians Universität, München Überblick: Molekulare Analysen 1) Generierung von Sequenzdaten: a) Sequenzierung