Alignments & Datenbanksuchen
|
|
|
- Hansi Goldschmidt
- vor 6 Jahren
- Abrufe
Transkript

1 WS2016/2017 F1-Praktikum Genomforschung und Sequenzanalyse: Einführung in Methoden der Bioinformatik Thomas Hankeln Alignments & Datenbanksuchen 1
2 Wiederholung Alignments Dynamic Programming Needleman-Wunsch: globales Alignment Smith-Waterman: lokales Alignment optimal Scoring-Matrizen: PAM & BLOSUM Gap penalties Dotplots: Visualisierung von Alignments FASTA, BLAST, BLAT heuristisch 2
3 Dynamic Programming Wie finde ich das beste alignment? Alle ausprobieren? Zu langsam: 2 x 300 Bp = Möglichkeiten Dynamic programming: löse kleine Sub-Probleme und konstruiere daraus Gesamtlösung 3
4 Dynamic Programming Zwei Sequenzen s und t der Länge L s und L t s = s1, s2, s3...sl s t = t1, t2, t3...tl t Wir möchte das optimale Alignment über die volle Länge Konstruiere Sub-Alignments Kombiniere diese Teillösungen rekursiv Benutze dabei eine dynamic programming-matrix M 4
5 Die DP Matrix t Eintrag M (i,j): Optimaler Score für Alignment von s mit t 0... Startfeld: Alignment von Nix mit Nix (= 0) s???????... 5
6 Füllen der Matrix: Schritt1 t einen Eintrag haben wir nun schon... von da aus gibt es drei Wege, sich durch die Matrix zu bewegen und weitere Felder auszufüllen s G A A T C G... 0 A T T C G C G C 6
7 Füllen der Matrix wir habe drei Möglichkeiten in jedem Matrixfeld Aligne Nt in s mit Nt in t 2. Aligne Nt in s mit Lücke in t 3. Aligne Lücke in s mit Nt in t (4. Aligne Lücke mit Lücke = Quatsch) 1...X X X X... 7
8 Füllen der Matrix: gap penalty > gap penalty einführen (z. B. -1) 1...X X X X... 8
9 Füllen der Matrix: matches Ziel: Maximaler Score Identitäts-Scores: Matches = +1, Mismatches = 0 9
10 Füllen der Matrix: Schritt 2 t gap penalty = -1 obere Reihe: aligne t mit gaps in s linke Spalte: aligne s mit gaps in t s G A A T C G G A T T C G C C -8 10
11 Füllen der Matrix für jedes Matrixfeld die drei Möglichkeiten berechnen: 1. Aligne beide Nts und addiere score (+1 oder 0) 2. Inseriere gap in s und addiere Inseriere gap in t und addiere -1 Das Ganze rekursiv auf das jeweilige Nachbarfeld beziehen: M (i-1, j-1) + score (a,b) M(i,j) = max M (i-1, j) + (-1) M (i, j-1) + (-1) 11
12 Füllen der Matrix Nur nochmal eine andere Formalisierung des Gesagten... 12
13 Füllen der Matrix M (i-1, j-1) + score (a,b) M(i,j) = max M (i-1, j) + (-1) M (i, j-1) + (-1) 0 G = 0 max -1 + (-1) = -2 A (-1) = -2 A -3 max T -4 C = (-1) = (-1) = -1 G G C A T T C G C
14 Aufgabe: Füllen der Matrix 0 A T T C G C Bestimme den optimalen Pfad des Alignments und Schreibe das Alignment Achtung: es kann mehrere gleichwertig optimale Alignments geben! G A A T C G G C -8 14
15 NW- Globales Alignment Alle möglichen optimalen Alignments werden durch trace-back als Pfad in der Matrix gefunden Sequenzen sind von Anfang bis Ende alignt. 15


16 Die Lösung match Gap in s Gap in t 16

17 NW- Globales Alignment 17
18 Global vs. Lokal 1 AGGATTGGAATGCTCAGAAGCAGCTAAAGCGTGTATGCAGGATTGGAATTAAAGAGGAGGTAGACCG AGGATTGGAATGCTAGGCTTGATTGCCTACCTGTAGCCACATCAGAAGCACTAAAGCGTCAGCGAGACCG TCAGAAGCAGCTAAAGCGT 42 TCAGAAGCA.CTAAAGCGT 1 AGGATTGGAATGCT 1 AGGATTGGAATGCT 39 AGGATTGGAAT 1 AGGATTGGAAT 62 AGACCG 66 AGACCG 18

19 Lokales Alignment Wenn die Sequenzen divergenter sind als zuvor... nur kurze konservierte Bereiche Abschnitte un-alignierbarer Sequenzen globales A. unsinnig Verwende Smith-Waterman lokales Alignment:...wie Reset-Knopf: starte erneut wenn Score unter Null fällt 19
20 Lokales Alignment lokales Alignment kann überall, auch innerhalb der Matrix starten obere Zeile und linke Spalte, sowie alle negativen Positionen werden mit 0 gefüllt trace-back vom Feld mit höchstem Score zuerst starten bei mehreren Startstellen: mehrere lokal konservierte Abschnitte 20
21 Lokales Alignment optimale lokale Alignments: TCG TCG A-TCGGC ATTC-GC TCGGC TC-GC AT-CGGC ATTCG-C TCGGC TCG-C G A A T C G G C A T T C G C
22 Zur Erinnerung... Optimales alignment heißt: höchster score, gegeben die Matrix und die gap penalty dies ist nicht unbedingt das biologisch sinnvollste Alignment Paarweise Alignment-tools produzieren immer etwas, manchmal auch Sinnloses... DNA: Paarweise alignment-tools können nur Nt-matches finden, wenn der gleiche Strang zweier Sequenzen verglichen wird 22
 W = - ( G open + (G ext x (n-1)) lin")
23 Gap Penalty linear: affin: W = - ( G open x Länge n ) W = - ( G open + (G ext x (n-1)) lin aff 23
24 Gap Penalty affin: W = G open + (G ext x ( n -1)) mit G ext < G open werden weniger aber, grössere Lücken favorisiert Werte zu gross: keine Lücken; Werte zu klein: zu viele Lücken gap penalties sind auf scoring-matrix abgestimmt für overlap alignment: end-gaps nicht bestraft 24
25 Scoring-Matrizen 4 x 4 für DNA 20 x 20 für Proteine Belohnungsscores für konservierte Positionen PAM (percent accepted mutation) BLOSUM (blocks substitution matrix) 25
26 PAM-Familie 1 PAM = 1 % Austausch (ca. 10 Mio Jahre) Was bedeutet dann PAM 250? umfasst multiple Austausche A > B > A PAM (klein) für ähnliche Sequenzen PAM (groß) für divergente Sequenzen 26
27 PAM 1 etc. 1. Finde sehr ähnliche Sequenzen (1% Divergenz) 2. Mache globales alignment (per Hand, 71 Gruppen) 3. Zähle die Austausche (1572) 4. Berechne die Scorewerte für die einzelnen Austausche 5. PAM2 = PAM1 x PAM1, PAM3 = PAM1 x PAM2 etc Extrapolation für größere Divergenzen durch Multiplikation 27
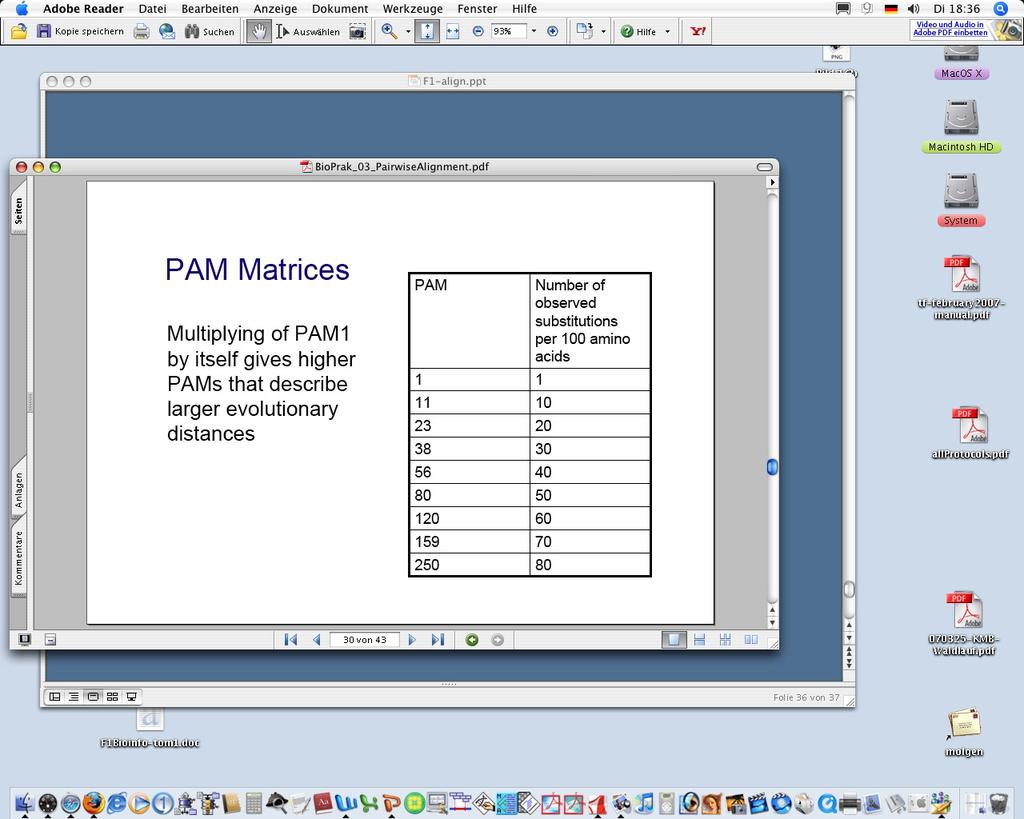
28 PAM 28
29 BLOSUM 1. Suche alignbare Blöcke ohne gaps (2000 Blöcke, 500 Proteinfamilien) 2. Zähle Austausche direkt, kalkuliere log odds-werte Log (a,b) / (a x b) 3. Mache dies ohne Extrapolation für Blöcke unterschiedlichen Grades an Ähnlichkeit! BLOSUM-Matrizen für unterschiedliche phylogenetische Abstände 29

 Gap penalty-scores!")
30 BLOSUM vs. PAM PAM 60 für 60% ähnliche Proteine 80 50% % Achtung: die meisten Matrizen in Vergleichsprogrammen haben assoziierte (und oft auch optimierte) Gap penalty-scores! Vorsicht bei drastischen Änderungen der gap penalty-werte relativ zu den Substitutions-Scores. 30

31 Matrizen und gap penalties bestimmen Signifikanz-Scores Wichtig im Grenzbereich... 31
32 Dot Plot manchmal reicht es, Alignments visuell zu vereinfachen... Sequenz mit sich selbst vergleichen: > Hauptdiagonale > parallele Diagonalen: direct repeats > orthogonale Diagonalen: inverted repeats > Quadrate mit noise : simple repeats 32
33 Dot Plot G A C C A G G A C G x x x A x x x C x x x C x x x A x x x G x x x G x x x A x x x C x x x
34 Dot Plot Mache einen Dotplot.. window =1 Stringenz = 1 window = 2 stringenz = 2 G A A T C G G A T T C G C... C 34
35 Dot Plot Mache einen Dotplot.. window = 2 stringenz = 2 G A A T C G G A T T C G C... C 35
36 Alignment für DB-Suchen Heuristische Algorithmen: gut & schnell, aber nicht unbedingt optimal seed-and extend : Suchsequenz in kurze Abschnitte ( words bzw. k-tuple ) aufbrechen (Wilbur und Lipman, 1983). zunächst sehr schnell nach word hits in der DB suchen diese dann erweitern zu längeren Segmenten 36
37 FastA Fast-All: Pearson & Lipman 1985 ist DB-Suchprogramm-Kollektion, aber auch Dateiformat Ktup-Suche: aa = 2, Nt = 6 Achtung: je länger das Ktup oder word, desto schneller, aber auch unsensitiver ist die Suche verbindet Ktuples, die nahe zueinander auf der gleichen Diagonale liegen 37
38 FastA 1. Bereche Index mit den Positionen aller Ktups in der Query-Sequenz 2. Berechne Index für Ktup- Positionen in der DB (einmal nur zu tun) 38 Folie: Tal Dagan, D dorf
39 FastA Ktup matches zwischen Query und EINER DB-Sequenz Init1-Region Diagonalen schlechter als Threshhold 3. Identifiziere Position passender Ktups zwischen Query und allen DB-Einträgen unter Verwendung der Indices 4. Identifiziere die 10 besten Diagonalen durch scoring: Höchster score = init1 39
 in Streifen um Diagonale")
40 FastA 5. Verbinde high-score- Diagonalen (ergibt initn-score) 6. Für höchste initn-matches kalkuliere optimales lokales Aligment (SW) in Streifen um Diagonale (Breite meist 32 As) (ergibt opt-score) 40
41 FastA-Programmfamilie Folie: Tal Dagan, D dorf 41
42 FastA-Output Folie: Tal Dagan, D dorf 42

43 Bill Pearson says... 43
44 BLAST zunächst wird nach kurzen lokal passenden Abschnitten ( words ) gesucht, dann versucht BLAST2.0, die Bereiche neben den matching words unter Einbeziehung von Lücken zu optimieren (word size W = 11 bei DNA) 44
45 BLAST Index- Einträge der Länge w Suchsequenz erster Hit Datenbanksequenz Gibt es 2.Hit? Fensterlänge A HSPs zwei lokale Alignments, Verknüpfung über Lücken falls möglich erlaubt 45
46 BLAST Parameter E-value: Wahrscheinlichkeit, dass ein solcher match zufällig in einer DB derselben Größe gefunden wird Filters: entfernt repetitive (low-complexity) Regionen Matrix: PAM, BLOSUM (machen manchmal den Unterschied!) Datenbanken: noch entscheidender!!! Vielfalt! Limits: z. B. nur in bestimmten Taxa suchen... Alignments und Descriptions: können bis zu Tausend angezeigt werden 46
47 Altschul et al. 1990, 1997 BLAST Programmfamilie schneller als FASTA! sucht lokale Alignments in DB ausgefeilte Such-Statistik (Karlin & Altschul 1990) Words können ähnlich sein (ungleich FastA) Low-complexity-Regionen werden entfernt 47
 blastx")
48 BLAST : Endecke die Möglichkeiten... blastn blastp DNA-Sequenz DNA-DB > nur nahe Verwandtschaft; beide Stränge verglichen As-Sequenz Protein-DB > entfernte Verwandtschaft (default:blosum62) blastx DNA-Seq > in 6 Leserahmen translatiert Protein-DB > findet mögliche Proteine in einer nichtcharakterisierten DNA-Sequenz (z.b. EST)! 48
 > findet nicht-annotierte Genregionen in DNA-DB- Sequenzen tblastx 6-frame-Translation")
")
49 BLAST : Endecke die Möglichkeiten... tblastn As-Seq gegen DNA-DB (6-frame translatiert!) > findet nicht-annotierte Genregionen in DNA-DB- Sequenzen tblastx 6-frame-Translation einer DNA-Seq 6-frame-Translation einer DNA-DB > Analyse von ESTs auf Proteinebene zur Detektion entfernter Verwandtschaft > kann nicht mit nr-db benutzt werden (zu aufwendig) 49
50 MegaBLAST sehr schnelle Nt-Suche (10x schneller als BLASTN) für sehr ähnliche Sequenzen word size : default 28 nicht-affine gap penalty: schneller, weniger Memory erforderlich > mehr, aber kürzere gaps Anwendung: schnelles Alignment zwischen ähnlichen Sequenzen, z. B. menschliche cdna an menschliches Genom alignen 50
 Suche nach discontiguous words besser")
51 Ma et al Discontiguous MegaBLAST schaut nicht -wie BLASTN und MegaBLAST- nach exakten word-matches (wird unproduktiv bei < 80 % id) Suche nach discontiguous words besser (weniger, aber signifikantere hits): Anwendung: schnelle Nt- Suche bei entfernt verwandten DNA-Sequenzen auf EST und Genomebene (z.b. mit Datenbank TRACES ) 51
52 BLAT BLAST-like alignment tool DNA-BLAT findet 40 Bp (>95% id) oder länger extrem schnell (500xBLAST) Protein-BLAT findet 20 aa (>80%id) Index (DNA) enthält alle nicht-überlappenden 11-mere des Genoms (1 Gb RAM) Index wird gebraucht um passende Regionen im Genom schnell zu identifizieren, die dann für genaueren Vergleich hochgeladen werden Anwendung: lokales Alignment zwischen längeren Sequenzen, z. B. cdna an menschliches Genom alignen 52
53 Score-Statistik BLAST berechnet Signifikanz aus Simulationen mit normalen, d. h. durchschnittlichen Sequenzen FASTA erstellt Verteilung von similarity scores während der DB-Suche (selektiert scores aus DB mit realen Sequenzen) PRSS (aus FASTA-Paket) berechnet Signifikanz durch Erstellen Hunderter von shuffled (random) sequences gleicher Länge und Zusammensetzung 53
54 Score-Statistik Reale unverwandte Sequenzen haben similarity scores wie zufällige Sequenzen Wenn die Similarität statistisch signifikant nicht ZUFÄLLIG ist, muss sie daher auf VERWANDSCHAFT schließen lassen E-Values < sind erfahrungsgemäß Treffer 54

55 DNA vs. Protein 55

56 Was ist besser? ssearch Sensitivität Speed 56

57 Bill Pearson says... 57

58 Bill Pearson says... 58
59 Anhang 59


60 Dotlet Score
 61")
61 X Inverted Repeat (Foldback Transposon) 61
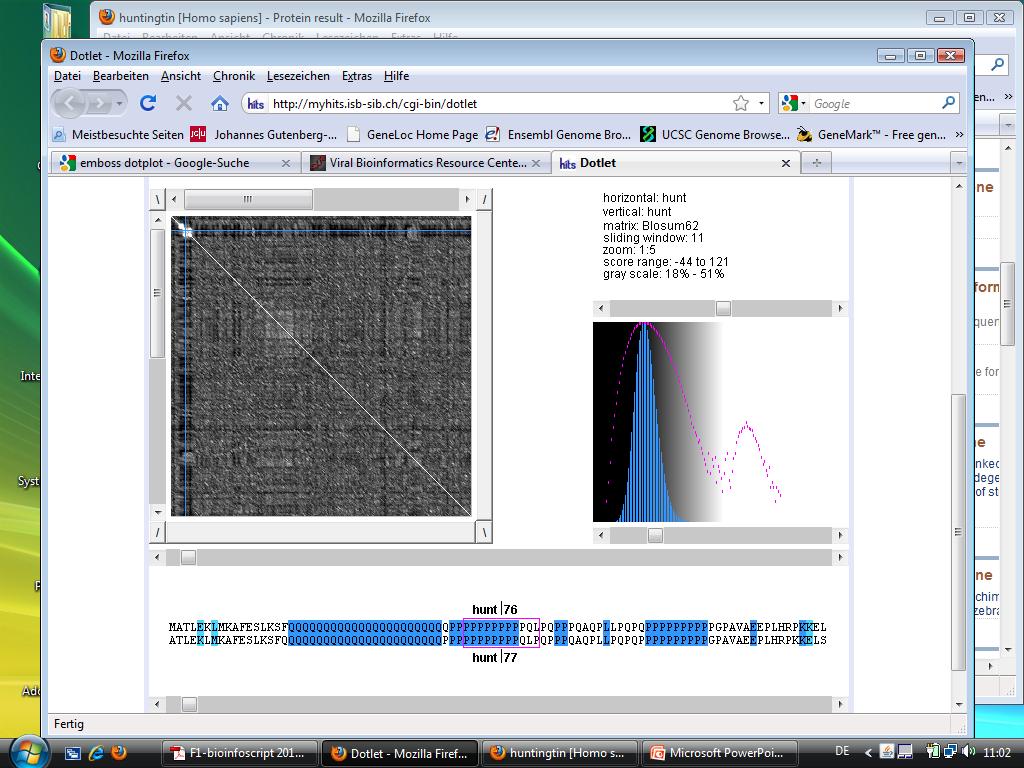
62 Huntingtin 62
63 63

64 HMG-SRY 64

65 Welches Tool auf der EMBOSS-Seite halten Sie intuitiv für geeignet?
66 Needleman-Wunsch Alignment Standard Einstellungen Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EBLOSUM62 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 158 # Identity: 68/158 (43.0%) # Similarity: 91/158 (57.6%) # Gaps: 19/158 (12.0%) # Score: # # #======================================= EMBOSS_ APLSADQASLVKSTWAQVRNSEVEILAAVFTAYPDIQ 37 :.... : :.: : EMBOSS_001 1 MKFIILALCVAAASALSGDQIGLVQSTYGKVKGDSVGILYAVFKADPTIQ 50 EMBOSS_ ARFPQFAGKDVASIKDTGAFATHAGRIVGFVSEIIALIGNESNAPAVQTL 87.. :.:... : :..:.:..:... EMBOSS_ AAFPQFVGKDLDAIKGGAEFSTHAGRIVGFLGGVI------DDLPNIGKH 94 EMBOSS_ VGQLAASHKARGISQAQFNEFRAGLVSYVSSNVAWNAAAESAWTAGLDNI :. ::....:: :..:.:.. :..... EMBOSS_ VDALVATHKPRGVTHAQFNNFRAAFIAYLKGHVDYTAAVEAAWGATFDAF 144 EMBOSS_ FGLLFAAL 145.:.: EMBOSS_ FGAVFAKM 152
67 Erniedrigen Sie einmal drastisch die gap penalty-werte ( z.b. von 10 auf 1) # Aligned_sequences: 2 Was # 1: EMBOSS_001 passiert? # 2: EMBOSS_001 # Matrix: EBLOSUM62 # Gap_penalty: 1.0 # Extend_penalty: 0.0 # # Length: 168 # Identity: 77/168 (45.8%) # Similarity: 99/168 (58.9%) # Gaps: 39/168 (23.2%) # Score: # # #======================================= EMBOSS_ APL-----SA---DQASLVKSTWAQVR-NSEVEILAAVFTAY-P 34.. : :.: : :... EMBOSS_001 1 MKFIILA-LCVAAASALSGDQIGLVQSTYGKVKGDS-VGILYAVFKA-DP 47 EMBOSS_ DIQARFPQFAGKDV-ASIKDTGA-FATHAGRIVGFVSEIIA-L--IGNES :. : :..:. : EMBOSS_ TIQAAFPQFVGKDLDA-IKG-GAEFSTHAGRIVGFLGGVIDDLPNIG-K- 93 EMBOSS_ NAPAVQTLVGQLAASHKARGISQAQFNEFRAGLVSYVSSNVAWNAAAESA 129 :.. :. ::....:: :..:.:.. : EMBOSS_ H---VDALV----ATHKPRGVTHAQFNNFRAAFIAYLKGHVDYTAAVEAA 136 EMBOSS_ WTAG--LDNIFGLLFAAL :.: EMBOSS_ W--GATFDAFFGAVFAKM 152
68 Probieren Sie den alternativ angebotenen Alignment- Algorithmus aus: was ändert sich? lokales S-W Alignment # Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EBLOSUM62 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 143 # Identity: 68/143 (47.6%) # Similarity: 90/143 (62.9%) # Gaps: 6/143 ( 4.2%) # Score: # # #======================================= EMBOSS_001 3 LSADQASLVKSTWAQVRNSEVEILAAVFTAYPDIQARFPQFAGKDVASIK : :.: : :.: EMBOSS_ LSGDQIGLVQSTYGKVKGDSVGILYAVFKADPTIQAAFPQFVGKDLDAIK 65 EMBOSS_ DTGAFATHAGRIVGFVSEIIALIGNESNAPAVQTLVGQLAASHKARGISQ : :..:.:..: :. ::. EMBOSS_ GGAEFSTHAGRIVGFLGGVI------DDLPNIGKHVDALVATHKPRGVTH 109 EMBOSS_ AQFNEFRAGLVSYVSSNVAWNAAAESAWTAGLDNIFGLLFAAL :: :..:.:.. :......:.: EMBOSS_ AQFNNFRAAFIAYLKGHVDYTAAVEAAWGATFDAFFGAVFAKM 152
69 Hämoglobin-Unterinheiten Standard Einstellungen Globales Needleman-Wunsch Alignment Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EBLOSUM62 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 149 # Identity: 65/149 (43.6%) # Similarity: 90/149 (60.4%) # Gaps: 9/149 ( 6.0%) # Score: # # #======================================= EMBOSS_001 1 MV-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-D 48 :.: :... :..... :.:.:. :.:.. EMBOSS_001 1 MVHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGD 48 EMBOSS_ LS-----HGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLR 93. :.:....::.: : ::...:. :... EMBOSS_ LSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLH 98 EMBOSS_ VDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR 142. :.:. : :...:. :..... EMBOSS_ VDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH 14
70 Alignen Sie die Sequenzen der beiden Untereinheiten des Hämoglobins und erhöhen Sie einmal drastisch die gap penalty- Werte. Was ändert sich? Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EBLOSUM62 # Gap_penalty: # Extend_penalty: 10.0 # # Length: 147 # Identity: 44/147 (29.9%) # Similarity: 62/147 (42.2%) # Gaps: 5/147 ( 3.4%) # Score: # # #======================================= EMBOSS_ MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFP :.....:... EMBOSS_001 1 MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLS 50 EMBOSS_ HFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVD :.:....::.: : ::...:. :... EMBOSS_ TPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVD 100 EMBOSS_ PVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR 142. :.:. : :...:. :..... EMBOSS_ PENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH 147
71 Probieren Sie ein paarweises Alignment mit den folgenden zwei Sequenzen, die eine mrna und eine zu ihr passende microrna (mirna) darstellen. # Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EDNAFULL # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 24 # Identity: 18/24 (75.0%) # Similarity: 18/24 (75.0%) # Gaps: 3/24 (12.5%) # Score: 48.0 # # #======================================= EMBOSS_001 1 ACUA-CCUGCACUGUA-AGCACUU EMBOSS_ ACUAGCAUCCAC-AUAGAGCACUU 46
72 Dme glob1 Dme glob2 NH2-...ekFpf...raHag...vsHip...-COOH NH2-...nfFrk...hgHam...ptHlk...-COOH EMBOSS_ mns 3.:. EMBOSS_001 1 msqisklthisrisqnnqsdgsdedkfrranfpvypkplpdrdlsykade 50 EMBOSS_001 4 devqlikk-----tweipvatptdsgaailtqffnrf-psnlekfpfrd- 46 :..:::.. :: : EMBOSS_ neftmvekaslrnawr liepfqrrfgkenfysfltrne 88 EMBOSS_ vpleelsgnarfrahagriirvfdesiqvlgqdgdle-- 83 :..:...::::..:.:..:. EMBOSS_ dlinffrkdgkinlsklhg------hamammklmsklvqtl--dcnlafr 130 EMBOSS_ kldeiwtkiavshiprtvskesynqlkgvildvlta acsld : :...:..:......:...:. :. EMBOSS_ lalde----nlpthlkngidpdymrmlatalksyilassvienhnscsls 176 EMBOSS_ esqaatwaklvdhv--ygiifka idddgnak : :..::.: :... EMBOSS_ ng----larlveivgeyavvdearkramstalrttvddagnrivkvalgt 222 # Matrix: EBLOSUM62 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 250 # Identity: 37/250 (14.8%) # Similarity: 67/250 (26.8%) # Gaps: 125/250 (50.0%) # Score: 39.5
73 Welche Substitutionsmatrix würden Sie anstatt der default-matrix wählen? Wechseln Sie auf eine besser geeignete Matrix! Notieren Sie die Werte! Wird das Alignment besser? # Matrix: EBLOSUM45 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 239 # Identity: 37/239 (15.5%) # Similarity: 69/239 (28.9%) # Gaps: 103/239 (43.1%) # Score: # # #======================================= Dme glob1 Dme glob2 NH2-...ekFpf...raHag...vsHip...-COOH NH2-...nfFrk...hgHam...ptHlk...-COOH EMBOSS_ mns 3.:. EMBOSS_001 1 msqisklthisrisqnnqsdgsdedkfrranfpvypkplpdrdlsykade 50 EMBOSS_001 4 devqlikk-----tweipvatptdsgaailtqffnrfpsnlekfpfrd-- 46 :..:::..:......:.. :...:....: EMBOSS_ neftmvekaslrnawrliepfqrrfgkenfysfltr-nedlinffrkdgk 99 EMBOSS_ vpleelsgnarfrahagriirvfdesiqvlgqdgdle---kldeiwtkia 93 :..:...::::..:.:..:..... EMBOSS_ inlsklhg------hamammklmsklvqtl--dcnlafrlaldenlp EMBOSS_ vshiprtvskesynqlkgvildvlta acsldesqaatwaklv 135 : :...:..:......:...:. :.... : EMBOSS_ thlkngidpdymrmlatalksyilassvienhnscslsng----larlv 183 EMBOSS_ dhv--ygiifka idddgnak :..::.: :... EMBOSS_ eivgeyavvdearkramstalrttvddagnrivkvalgt 222
74 Ändern Sie nun die gap extension penalty schrittweise zunächst auf 1.0, dann 5.0. Betrachten Sie den letzten Fall: Erfüllt das alignment nun besser die strukturbiologischen Vorgaben? # Aligned_sequences: 2 # 1: EMBOSS_001 # 2: EMBOSS_001 # Matrix: EBLOSUM45 # Gap_penalty: 10.0 # Extend_penalty: 5.0 # # Length: 230 # Identity: 23/230 (10.0%) # Similarity: 59/230 (25.7%) # Gaps: 85/230 (37.0%) # Score: 44.0 # # #======================================= EMBOSS_ EMBOSS_001 1 msqisklthisrisqnnqsdgsdedkfrranfpvypkplpdrdlsykade 50 EMBOSS_ mnsdevqlikktweipvatptdsgaailtqffnrfpsnlekfpfrdvp ::...:......:.. :...:..... EMBOSS_ neftmvekaslrnawrliepfqrrfgkenfysfltr-nedlinf-frkdg 98 EMBOSS_ leelsgnarfrahagriirvfdesiqvlgqdgdlekldeiwtkiavship ::....::::..:.:..:.:...:...: :. EMBOSS_ kinls---klhghamammklmsklvqtl--dcnl-afrlaldenlpthlk 142 EMBOSS_ rtvskesynqlkgvildvltaacsldesqaatwaklvdhvygiifk-aid 147..:..:......:...:. :..::...:.:.:..:..:.. :.: :. EMBOSS_ ngidpdymrmlatalksyilassvienhnscslsnglarlveivgeyavv 192 EMBOSS_ ddgnak :...: EMBOSS_ dearkramstalrttvddagnrivkvalgt 222
75 BLAST für s Laborleben Die Sequenz ist im kodierenden Bereich fehlerhaft: wo vermuten Sie Fehler? Human calmodulin mrna, complete cds Sequence ID: gb M HUMCAMLength: 1126Number of Matches: 1 Alignment statistics for match #1 Score Expect Identities Gaps Strand 819 bits(443) /450(99%) 2/450(0%) Plus/Plus Query 173 GCTGACCAACTGACTGAAGAGCAGATTGCAGAATTCAAAGAAGCTTTTTCATTATTTGAC 232 Sbjct 56 GCTGACCAACTGACTGAAGAGCAGATTGCAGAATTCAAAGAAGCTTTTTCATTATTTGAC 115 Query 233 AAAGATGGTGATGGCACTATAACAACAAAGGAACTTGGGACTGTAATGAGATCTCTTGGG 292 Sbjct 116 AAAGATGGTGATGGCACTATAACAACAAAGGAACTTGGGACTGTAATGAGATCTCTTGGG 175 Query 293 CAGAATCCCACAGAAGCAGAGTTACAGGACATGATTAATGAAGTAGATGCTGATGGTAAT 352 Sbjct 176 CAGAATCCCACAGAAGCAGAGTTACAGGACATGATTAATGAAGTAGATGCTGATGGTAAT 235 Query 353 GGCACAATTGACTTTCCTGAATTTCTGACAATGATGGCAAG-AAAATGAAAGACACAGAC 411 Del>framshift Sbjct 236 GGCACAATTGACTTTCCTGAATTTCTGACAATGATGGCAAGAAAAATGAAAGACACAGAC 295 Query 412 AGTGAAGAAGAAATTAGAGAAGCATTCCGTGTGTTTGACAAGGATGGCAATGGCTATATT 471 Sbjct 296 AGTGAAGAAGAAATTAGAGAAGCATTCCGTGTGTTTGACAAGGATGGCAATGGCTATATT 355 Query 472 AGTGCTGCAGAACTTCGCCATGTGATGACAAACCTTGGAGAGAAGTTAACAGATGAAGAA 531 Sbjct 356 AGTGCTGCAGAACTTCGCCATGTGATGACAAACCTTGGAGAGAAGTTAACAGATGAAGAA 415 Query 532 GTTGATGAAAATGATCAGGGAAGCAGATATTGATGGTGATGGTCAAGTAAACTATGAAGA 591ins>Frameshift weg Sbjct 416 GTTGATG-AAATGATCAGGGAAGCAGATATTGATGGTGATGGTCAAGTAAACTATGAAGA 474 Query 592 GTTTGTACAAATGATGACAGCAAAGTGAAG 621 Sbjct 475 GTTTGTACAAATGATGACAGCAAAGTGAAG 504
76 BLAST für s Laborleben Die Sequenz ist im kodierenden Bereich fehlerhaft: wo vermuten Sie Fehler? RecName: Full=Calmodulin; Short=CaM [Rattus norvegicus] Sequence ID: sp P CALM_RATLength: 149Number of Matches: 1 Alignment statistics for match #1 Score Expect Method Identities Positives Gaps Frame 194 bits(493) 1e-60 Compositional matrix adjust. 102/148(69%) 113/148(76%) 0/148(0%) +2 Query 173 ADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGN 352 ADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGN Sbjct 2 ADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGN 61 Query 353 GTIDFPEFLTMMARK*KTQTVKKKLEKHSVCLTRMAMAILVLQNFAM**QTLERS*QMKK 532 del>frameshift GTIDFPEFLTMMARK K L ++ Sbjct 62 GTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLGEKLTDEE 121 Query 533 LMKMIREADIDGDGQVNYEEFVQMMTAK 616>ins>framshift weg + +MIREADIDGDGQVNYEEFVQMMTAK Sbjct 122 VDEMIREADIDGDGQVNYEEFVQMMTAK 149
Alignments & Datenbanksuchen
 WS2015/2016 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignments & Datenbanksuchen 1 break-thru Doolittle et al. 1983, Waterfield et al. 1983 > DB-Suche...
WS2015/2016 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignments & Datenbanksuchen 1 break-thru Doolittle et al. 1983, Waterfield et al. 1983 > DB-Suche...
Alignments & Datenbanksuchen
 WS2017/2018 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignments & Datenbanksuchen 1 break-thru Doolittle et al. 1983, Waterfield et al. 1983 > DB-Suche...
WS2017/2018 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignments & Datenbanksuchen 1 break-thru Doolittle et al. 1983, Waterfield et al. 1983 > DB-Suche...
Bioinformatik. BLAST Basic Local Alignment Search Tool. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik BLAST Basic Local Alignment Search Tool Ulf Leser Wissensmanagement in der Bioinformatik Heuristische Alignierung Annotation neuer Sequenzen basiert auf Suche nach homologen Sequenzen in
Bioinformatik BLAST Basic Local Alignment Search Tool Ulf Leser Wissensmanagement in der Bioinformatik Heuristische Alignierung Annotation neuer Sequenzen basiert auf Suche nach homologen Sequenzen in
Alignment von DNA- und Proteinsequenzen
 WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
Proseminar Bioinformatik
 Proseminar Bioinformatik Thema Algorithmic Concepsts for Searching in Biological Databases von Uwe Altermann 30.05.2009 1 Einführung Im Folgenden soll ein Überblick über die verschiedenen algorithmischen
Proseminar Bioinformatik Thema Algorithmic Concepsts for Searching in Biological Databases von Uwe Altermann 30.05.2009 1 Einführung Im Folgenden soll ein Überblick über die verschiedenen algorithmischen
MBI: Sequenz-Vergleich mit Alignment
 MBI: Sequenz-Vergleich mit Alignment Bernhard Haubold 28. Oktober 2014 Wiederholung: Was ist Bioinformatik? Historische Übersicht; CABIOS Bioinformatics Gemeinsames Thema: Information in vivo DNA Epigenetik
MBI: Sequenz-Vergleich mit Alignment Bernhard Haubold 28. Oktober 2014 Wiederholung: Was ist Bioinformatik? Historische Übersicht; CABIOS Bioinformatics Gemeinsames Thema: Information in vivo DNA Epigenetik
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 06. Paarweises Alignment Teil II Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 06. Paarweises Alignment Teil II Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Aufgabenblatt 5. Silke Trißl Wissensmanagement in der Bioinformatik
 Aufgabenblatt 5 Silke Trißl Wissensmanagement in der Bioinformatik Zuerst! FRAGEN? Silke Trißl: Bioinformatik für Biophysiker 2 Exercise 1 + 2 Modify program to compare protein sequence read substitution
Aufgabenblatt 5 Silke Trißl Wissensmanagement in der Bioinformatik Zuerst! FRAGEN? Silke Trißl: Bioinformatik für Biophysiker 2 Exercise 1 + 2 Modify program to compare protein sequence read substitution
MOL.504 Analyse von DNA- und Proteinsequenzen. Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
Gleichheit, Ähnlichkeit, Homologie
 Gleichheit, Ähnlichkeit, Homologie Identität (identity) Verhältnis der Anzahl identischer Aminosäuren zur Gesamtzahl der Aminosäuren; objektiv Ähnlichkeit (similarity) Verhältnis ähnlicher Aminosäuren
Gleichheit, Ähnlichkeit, Homologie Identität (identity) Verhältnis der Anzahl identischer Aminosäuren zur Gesamtzahl der Aminosäuren; objektiv Ähnlichkeit (similarity) Verhältnis ähnlicher Aminosäuren
Dot-Matrix Methode. (Java) (Javascript) 80
 (Javascript) 80") Dot-Matrix Methode Vergleich zweier Sequenzen (DNA oder Aminosäuren) Idee: gleiche Basen (Aminosäuren) in x-y Diagramm markieren Sequenz 1: ADRWLVKQN Sequenz 2: ADKFIVRDE http://myhits.vital-it.ch/cgi-bin/dotlet
Dot-Matrix Methode Vergleich zweier Sequenzen (DNA oder Aminosäuren) Idee: gleiche Basen (Aminosäuren) in x-y Diagramm markieren Sequenz 1: ADRWLVKQN Sequenz 2: ADKFIVRDE http://myhits.vital-it.ch/cgi-bin/dotlet
Zentrum für Bioinformatik. Übung 4: Revision. Beispielfragen zur Klausur im Modul Angewandte Bioinformatik (erste Semesterhälfte)
") Andrew Torda Björn Hansen Iryna Bondarenko Zentrum für Bioinformatik Übung zur Vorlesung Angewandte Bioinformatik Sommersemester 2014 20./23.06.2014 Übung 4: Revision Beispielfragen zur Klausur im Modul
Andrew Torda Björn Hansen Iryna Bondarenko Zentrum für Bioinformatik Übung zur Vorlesung Angewandte Bioinformatik Sommersemester 2014 20./23.06.2014 Übung 4: Revision Beispielfragen zur Klausur im Modul
Softwarewerkzeuge der Bioinformatik
 Bioinformatik Wintersemester 2006/2007 Tutorial 2: paarweise Sequenzaligments BLAST Tutorial 2: BLAST 1/22 Alignment Ausrichten zweier oder mehrerer Sequenzen, um: ihre Ähnlichkeit quantitativ zu erfassen
Bioinformatik Wintersemester 2006/2007 Tutorial 2: paarweise Sequenzaligments BLAST Tutorial 2: BLAST 1/22 Alignment Ausrichten zweier oder mehrerer Sequenzen, um: ihre Ähnlichkeit quantitativ zu erfassen
Vorlesung Einführung in die Bioinformatik
 Vorlesung Einführung in die Bioinformatik Dr. Stephan Weise 04.04.2016 Einführung Gegeben: zwei Sequenzen Gesucht: Ähnlichkeit quantitativ erfassen Entsprechungen zwischen einzelnen Bausteinen beider Sequenzen
Vorlesung Einführung in die Bioinformatik Dr. Stephan Weise 04.04.2016 Einführung Gegeben: zwei Sequenzen Gesucht: Ähnlichkeit quantitativ erfassen Entsprechungen zwischen einzelnen Bausteinen beider Sequenzen
Bioinformatik für Biochemiker
 Bioinformatik für Biochemiker Oliver Kohlbacher W 2009/2010 7. Datenbanksuche Abt. imulation biologischer ysteme WI/ZBIT, Eberhard Karls Universität Tübingen Übersicht Datenbanksuche statt Alignment Heuristiken:
Bioinformatik für Biochemiker Oliver Kohlbacher W 2009/2010 7. Datenbanksuche Abt. imulation biologischer ysteme WI/ZBIT, Eberhard Karls Universität Tübingen Übersicht Datenbanksuche statt Alignment Heuristiken:
BLAST. Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02
 BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
Was ist Bioinformatik?
 9. Kurstag: Bioinformatik Der Begriff "Bioinformatik" wurde 1989 erstmals von D.R. Masys im JOURNAL OF RESEARCH OF THE NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY erwähnt. Was ist Bioinformatik? Die
9. Kurstag: Bioinformatik Der Begriff "Bioinformatik" wurde 1989 erstmals von D.R. Masys im JOURNAL OF RESEARCH OF THE NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY erwähnt. Was ist Bioinformatik? Die
Primärstruktur. Wintersemester 2011/12. Peter Güntert
 Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Primärstruktur Wintersemester 2011/12 Peter Güntert Primärstruktur Beziehung Sequenz Struktur Proteinsequenzen, Sequenzdatenbanken Sequenzvergleich (sequence alignment) Sequenzidentität, Sequenzhomologie
Sequenzvergleich und Datenbanksuche
 Sequenzvergleich und Datenbanksuche Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann
Sequenzvergleich und Datenbanksuche Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann
Algorithmische Anwendungen WS 2005/2006
 Algorithmische Anwendungen WS 2005/2006 Sequenzalignment Gruppe F_lila_Ala0506 Allal Kharaz Yassine ELassad Inhaltsverzeichnis 1 Problemstellungen...................................... 3 1.1 Rechtschreibkorrektur...............................
Algorithmische Anwendungen WS 2005/2006 Sequenzalignment Gruppe F_lila_Ala0506 Allal Kharaz Yassine ELassad Inhaltsverzeichnis 1 Problemstellungen...................................... 3 1.1 Rechtschreibkorrektur...............................
Bioinformatik. Lokale Alignierung Gapkosten. Silke Trißl / Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
Multiple Alignments. Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann. Webseite zur Vorlesung
 Multiple Alignments Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at-
Multiple Alignments Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at-
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag
 WS2013/2014 Woche 3 - Montag") VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
Algorithmen für paarweise Sequenz-Alignments. Katharina Hembach
 Proseminar Bioinformatik WS 2010/11 Algorithmen für paarweise Sequenz-Alignments Katharina Hembach 06.12.2010 1 Einleitung Paarweise Sequenz-Alignments spielen in der Bioinformatik eine wichtige Rolle.
Proseminar Bioinformatik WS 2010/11 Algorithmen für paarweise Sequenz-Alignments Katharina Hembach 06.12.2010 1 Einleitung Paarweise Sequenz-Alignments spielen in der Bioinformatik eine wichtige Rolle.
Sequenz Alignment Teil 2
 Sequenz Alignment Teil 2 14.11.03 Vorlesung Bioinformatik 1 Molekulare Biotechnologie Dr. Rainer König Besonderen Dank an Mark van der Linden, Mechthilde Falkenhahn und der Husar Biocomputing Service Gruppe
Sequenz Alignment Teil 2 14.11.03 Vorlesung Bioinformatik 1 Molekulare Biotechnologie Dr. Rainer König Besonderen Dank an Mark van der Linden, Mechthilde Falkenhahn und der Husar Biocomputing Service Gruppe
Ihre Namen: Gruppe: Öffnen Sie die Fasta-Dateien nur mit einem Texteditor, z.b. Wordpad oder Notepad, nicht mit Microsoft Word oder Libre Office.
 Ihre Namen: Gruppe: Evolutionsbiologie 2, WS2016/2017: Bioinformatik - Übung 1 Erstellen Sie vor Beginn der Übung einen Ordner auf dem Desktop, in dem Sie alle benötigten Dateien speichern kö nnen (z.b.
Ihre Namen: Gruppe: Evolutionsbiologie 2, WS2016/2017: Bioinformatik - Übung 1 Erstellen Sie vor Beginn der Übung einen Ordner auf dem Desktop, in dem Sie alle benötigten Dateien speichern kö nnen (z.b.
Paarweises Sequenzalignment
 Methoden des Sequenzalignments Paarweises Sequenzalignment Austauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Methoden des Sequenzalignments Paarweises Sequenzalignment Austauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Studiengang Informatik der FH Gießen-Friedberg. Sequenz-Alignment. Jan Schäfer. WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel
 Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
BIOINF 1910 Bioinforma1k für Lebenswissenscha;ler Oliver Kohlbacher Datenbanksuche Problem Beispiele
 BIOINF 1910 Bioinforma1k für Lebenswissenscha;ler Oliver Kohlbacher SS 2011 08. Datenbanksuche: BLAST und PSI- BLAST Datenbanksuche Problem Gegeben: eine Sequenz und eine Sequenzdatenbank Gesucht: ähnlichste
BIOINF 1910 Bioinforma1k für Lebenswissenscha;ler Oliver Kohlbacher SS 2011 08. Datenbanksuche: BLAST und PSI- BLAST Datenbanksuche Problem Gegeben: eine Sequenz und eine Sequenzdatenbank Gesucht: ähnlichste
Paarweises Sequenzalignment
 Methoden des Sequenzalignments Paarweises Sequenzalignment Áustauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Methoden des Sequenzalignments Paarweises Sequenzalignment Áustauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Bachelorarbeit. Paarweise und multiple Alignments mit TBLASTX und DIALIGN
 Georg-August-Universität Göttingen Zentrum für Informatik ISSN Nummer 1612-6793 ZFI-BM-2005-28 Bachelorarbeit im Studiengang Angewandte Informatik Paarweise und multiple Alignments mit TBLASTX und DIALIGN
Georg-August-Universität Göttingen Zentrum für Informatik ISSN Nummer 1612-6793 ZFI-BM-2005-28 Bachelorarbeit im Studiengang Angewandte Informatik Paarweise und multiple Alignments mit TBLASTX und DIALIGN
Paarweises Sequenzalignment
 Methoden des Sequenzalignments Paarweises Sequenzalignment Áustauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Methoden des Sequenzalignments Paarweises Sequenzalignment Áustauschmatrizen Bedeutsamkeit von Alignments BLAST, Algorithmus Parameter Ausgabe http://www.ncbi.nih.gov Diese Vorlesung lehnt sich eng an
Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments
 Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments Sven Rahmann Genominformatik Universitätsklinikum Essen Universität Duisburg-Essen Universitätsallianz Ruhr Einführung Bisher: Berechnung
Algorithmen auf Sequenzen Paarweiser Sequenzvergleich: Alignments Sven Rahmann Genominformatik Universitätsklinikum Essen Universität Duisburg-Essen Universitätsallianz Ruhr Einführung Bisher: Berechnung
Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS Yvonne Lichtblau/Johannes Starlinger
 Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS 2017 Yvonne Lichtblau/Johannes Starlinger Presentations Assignment 2 Yvonne Lichtblau Übungen Grundlagen der Bioinformatik SS 2017
Grundlagen der Bioinformatik Assignment 3: Hierarchical Clustering SS 2017 Yvonne Lichtblau/Johannes Starlinger Presentations Assignment 2 Yvonne Lichtblau Übungen Grundlagen der Bioinformatik SS 2017
Algorithmische Bioinformatik 1
 Algorithmische Bioinformatik 1 Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2009 Übersicht Paarweises
Algorithmische Bioinformatik 1 Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2009 Übersicht Paarweises
Bioinformatik. Substitutionsmatrizen BLAST. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Alignment-Verfahren zum Vergleich biologischer Sequenzen
 zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
Threading - Algorithmen
 Threading - Algorithmen Florian Lindemann 22.11.2007 Florian Lindemann () Threading - Algorithmen 22.11.2007 1 / 25 Gliederung 1 Prospect Scoring Function Algorithmus Weitere Eigenschaften Komplexität
Threading - Algorithmen Florian Lindemann 22.11.2007 Florian Lindemann () Threading - Algorithmen 22.11.2007 1 / 25 Gliederung 1 Prospect Scoring Function Algorithmus Weitere Eigenschaften Komplexität
MOL.504 Analyse von DNA- und Proteinsequenzen. Übungsaufgaben BLAST-Sequenzsuche und -vergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben BLAST-Sequenzsuche und -vergleiche Ü6a blastn und blastx Verwenden Sie die in Übung 3 (Datenbanken) gefundene yqjm-sequenz aus Bacillus subtilis
MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben BLAST-Sequenzsuche und -vergleiche Ü6a blastn und blastx Verwenden Sie die in Übung 3 (Datenbanken) gefundene yqjm-sequenz aus Bacillus subtilis
Bioinformatik Für Biophysiker
 Bioinformatik Für Biophysiker Wintersemester 2005 / 2006 Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Lehrstuhl seit 10/2002 Schwerpunkte Algorithmen der Bioinformatik
Bioinformatik Für Biophysiker Wintersemester 2005 / 2006 Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Lehrstuhl seit 10/2002 Schwerpunkte Algorithmen der Bioinformatik
Bioinformatische Suche nach pre-mirnas
 Bioinformatische Suche nach pre-mirnas Vorbereitung: Lesen Sie die Publikationen Meyers et al., 2008, Criteria for Annotation of Plant MicroRNAs und Thieme et al., 2011, SplamiR prediction of spliced mirnas
Bioinformatische Suche nach pre-mirnas Vorbereitung: Lesen Sie die Publikationen Meyers et al., 2008, Criteria for Annotation of Plant MicroRNAs und Thieme et al., 2011, SplamiR prediction of spliced mirnas
Bioinformatik BLAT QUASAR. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik BLAT QUASAR Ulf Leser Wissensmanagement in der Bioinformatik Exklusionsmethode BYP Alignment zweier Strings A,B dauert O(n*m) K-Band Algorithmus benötigt O(sn 2 -vn) für A = B Gutes Verfahren,
Bioinformatik BLAT QUASAR Ulf Leser Wissensmanagement in der Bioinformatik Exklusionsmethode BYP Alignment zweier Strings A,B dauert O(n*m) K-Band Algorithmus benötigt O(sn 2 -vn) für A = B Gutes Verfahren,
BCDS Seminar. Protein Tools
 BCDS Seminar Protein Tools Gliederung Nützliche Tools Three-/one-letter Amino Acids' Сodes RandSeq Random Protein Sequence Generator Protein Colourer ProtParam PeptideCutter ProtScale TMHMM Server 2.0
BCDS Seminar Protein Tools Gliederung Nützliche Tools Three-/one-letter Amino Acids' Сodes RandSeq Random Protein Sequence Generator Protein Colourer ProtParam PeptideCutter ProtScale TMHMM Server 2.0
Aufgabe 7: Distanzbasierte Phylogenie: Neighbor Joining. Stefan Kröger, Philippe Thomas Wissensmanagement in der Bioinformatik
 Aufgabe 7: Distanzbasierte Phylogenie: Neighbor Joining Stefan Kröger, Philippe Thomas Wissensmanagement in der Bioinformatik Daten Wir verwenden neue Daten Die müssen sie ausnahmsweise selber suchen DNA-Sequenzen
Aufgabe 7: Distanzbasierte Phylogenie: Neighbor Joining Stefan Kröger, Philippe Thomas Wissensmanagement in der Bioinformatik Daten Wir verwenden neue Daten Die müssen sie ausnahmsweise selber suchen DNA-Sequenzen
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
1/10. Ihre Namen: Gruppe: Evolutionsbiologie 2, WS2015/2016: Bioinformatik - Übung 1
 Ihre Namen: Gruppe: Evolutionsbiologie 2, WS2015/2016: Bioinformatik - Übung 1 Erstellen Sie bitte vor Beginn der Übung einen Ordner auf dem Desktop, in dem Sie alle benötigten Dateien speichern können
Ihre Namen: Gruppe: Evolutionsbiologie 2, WS2015/2016: Bioinformatik - Übung 1 Erstellen Sie bitte vor Beginn der Übung einen Ordner auf dem Desktop, in dem Sie alle benötigten Dateien speichern können
MikroRNA-Bioinformatik. Annalisa Marsico High-throughput Genomics, FU Berlin
 MikroRNA-Bioinformatik Annalisa Marsico High-throughput Genomics, FU Berlin 07.12.2016 Lernziele Einführung in die Welt der nicht-kodierenden RNAs Inbesondere mikrorna Molekulare Funktionen von micrornas
MikroRNA-Bioinformatik Annalisa Marsico High-throughput Genomics, FU Berlin 07.12.2016 Lernziele Einführung in die Welt der nicht-kodierenden RNAs Inbesondere mikrorna Molekulare Funktionen von micrornas
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse
 Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
BLAST. Datenbanksuche mit BLAST. Genomische Datenanalyse 10. Kapitel
 Datenbanksuche mit BLAST BLAST Genomische Datenanalyse 10. Kapitel http://www.ncbi.nlm.nih.gov/blast/ Statistische Fragen Datenbanksuche Query Kann die globale Sequenzähnlichkeit eine Zufallsfluktuation
Datenbanksuche mit BLAST BLAST Genomische Datenanalyse 10. Kapitel http://www.ncbi.nlm.nih.gov/blast/ Statistische Fragen Datenbanksuche Query Kann die globale Sequenzähnlichkeit eine Zufallsfluktuation
Bioinformatik Für Biophysiker
 Bioinformatik Für Biophysiker Sommersemester 2009 Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Schwerpunkte Algorithmen der Bioinformatik Management
Bioinformatik Für Biophysiker Sommersemester 2009 Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Wissensmanagement in der Bioinformatik Schwerpunkte Algorithmen der Bioinformatik Management
Algorithmen und Datenstrukturen in der Bioinformatik Erstes Übungsblatt WS 05/06 Musterlösung
 Konstantin Clemens Johanna Ploog Freie Universität Berlin Institut für Mathematik II Arbeitsgruppe für Mathematik in den Lebenswissenschaften Algorithmen und Datenstrukturen in der Bioinformatik Erstes
Konstantin Clemens Johanna Ploog Freie Universität Berlin Institut für Mathematik II Arbeitsgruppe für Mathematik in den Lebenswissenschaften Algorithmen und Datenstrukturen in der Bioinformatik Erstes
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2011 1. Was ist Bioinformatik? Ablauf und Formales Ringvorlesung
Einführung in die Bioinformatik Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de SS 2011 1. Was ist Bioinformatik? Ablauf und Formales Ringvorlesung
Biowissenschaftlich recherchieren
 Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
Vortrag 2: Proteinsequenzen und Substitutionsmatrizen
 Vortrag 2: Proteinsequenzen und Substitutionsmatrizen Was sind Proteinsequenzen? Die DNA-Forschung hat sich auf spezielle Abschnitte auf den Strängen der DNA-Moleküle konzentriert, den sog. Protein-codierenden
Vortrag 2: Proteinsequenzen und Substitutionsmatrizen Was sind Proteinsequenzen? Die DNA-Forschung hat sich auf spezielle Abschnitte auf den Strängen der DNA-Moleküle konzentriert, den sog. Protein-codierenden
AlgoBio WS 16/17 Protein-DNA Interaktionen ChiP-Seq Datenanalyse. Annalisa Marsico
 AlgoBio WS 16/17 Protein-DNA Interaktionen ChiP-Seq Datenanalyse Annalisa Marsico 6.02.2017 Protein-DNA Interaktionen Häufig binden sich Proteine an DNA, um ihre biologische Funktion zu regulieren. Transkriptionsfaktoren
AlgoBio WS 16/17 Protein-DNA Interaktionen ChiP-Seq Datenanalyse Annalisa Marsico 6.02.2017 Protein-DNA Interaktionen Häufig binden sich Proteine an DNA, um ihre biologische Funktion zu regulieren. Transkriptionsfaktoren
Kapitel 7: Sequenzen- Alignierung in der Bioinformatik
 Kapitel 7: Sequenzen- Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS11 19. VO 14. Juni 2007 1 Literatur für diese VO Volker
Kapitel 7: Sequenzen- Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS11 19. VO 14. Juni 2007 1 Literatur für diese VO Volker
FOLDALIGN und sein Algorithmus. Nadine Boley Silke Szymczak
 FOLDALIGN und sein Algorithmus Nadine Boley Silke Szymczak Gliederung 2 Einleitung Motivation des Ansatzes zu FOLDALIGN Sankoff-Algorithmus Globales Alignment Zuker-Algorithmus Kombination FOLDALIGN Algorithmus,
FOLDALIGN und sein Algorithmus Nadine Boley Silke Szymczak Gliederung 2 Einleitung Motivation des Ansatzes zu FOLDALIGN Sankoff-Algorithmus Globales Alignment Zuker-Algorithmus Kombination FOLDALIGN Algorithmus,
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 5. Paarweises Alignment Teil I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 5. Paarweises Alignment Teil I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Genvorhersage & Genom- Annotation
 WS2016/2017 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Genvorhersage & Genom- Annotation Ebenen der Annotation Genstruktur (Exons/Introns, UTR s, Promoter,
WS2016/2017 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Genvorhersage & Genom- Annotation Ebenen der Annotation Genstruktur (Exons/Introns, UTR s, Promoter,
Bioinformatik. Multiple String Alignment I. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Multiple String Alignment I Ulf Leser Wissensmanagement in der Bioinformatik BLAST2: Zwei-Hit-Strategie Original: Alle Hits mit Score > t werden zu MSPs verlängert Extensionen fressen >90%
Bioinformatik Multiple String Alignment I Ulf Leser Wissensmanagement in der Bioinformatik BLAST2: Zwei-Hit-Strategie Original: Alle Hits mit Score > t werden zu MSPs verlängert Extensionen fressen >90%
Datenbanken in der Bioinformatik
 Datenbanken in der Bioinformatik Kapitel 3 Sequenzierung und Genexpressionsanalyse http://dbs.uni-leipzig.de Institut für Informatik Vorläufiges Inhaltsverzeichnis 1. Grundlagen 2. Klassifizierung von
Datenbanken in der Bioinformatik Kapitel 3 Sequenzierung und Genexpressionsanalyse http://dbs.uni-leipzig.de Institut für Informatik Vorläufiges Inhaltsverzeichnis 1. Grundlagen 2. Klassifizierung von
Copyright, Page 1 of 5 Die Determinante
 wwwmathematik-netzde Copyright, Page 1 of 5 Die Determinante Determinanten sind ein äußerst wichtiges Instrument zur Untersuchung von Matrizen und linearen Abbildungen Außerhalb der linearen Algebra ist
wwwmathematik-netzde Copyright, Page 1 of 5 Die Determinante Determinanten sind ein äußerst wichtiges Instrument zur Untersuchung von Matrizen und linearen Abbildungen Außerhalb der linearen Algebra ist
Bioinformatik. Dynamische Programmierung. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Homologie und Sequenzähnlichkeit. Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Einführung in die Bioinformatik
 Einführung in die Bioinformatik Ringvorlesung Biologie Sommer 07 Burkhard Morgenstern Institut für Mikrobiologie und Genetik Abteilung für Bioinformatik Goldschmidtstr. 1 Online Materialien zur Ringvorlesung:
Einführung in die Bioinformatik Ringvorlesung Biologie Sommer 07 Burkhard Morgenstern Institut für Mikrobiologie und Genetik Abteilung für Bioinformatik Goldschmidtstr. 1 Online Materialien zur Ringvorlesung:
Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST
 Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST 10.06.2010 Prof. Dr. Sven Rahmann 1 Sequenzvergleich: Motivation Hat man die DNA-Sequenz eines Gens, die Aminosäuresequenz
Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST 10.06.2010 Prof. Dr. Sven Rahmann 1 Sequenzvergleich: Motivation Hat man die DNA-Sequenz eines Gens, die Aminosäuresequenz
Sequenzen-Alignierung in der Bioinformatik
 Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 2. VO 2.0.2006 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 2. VO 2.0.2006 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Fortgeschrittene Netzwerk- und Graph-Algorithmen
 Fortgeschrittene Netzwerk- und Graph-Algorithmen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Wintersemester
Fortgeschrittene Netzwerk- und Graph-Algorithmen Prof. Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Wintersemester
Protokoll von Johnnie Akgün
 Aufgabenstellung: Protokoll von Johnnie Akgün 0401350 Identifizierung von strukturierten non coding RNAs von 6 verschiedenen Aspergillus Stämmen. Arbeitsschritte: Begonnen wird mit einem Fasta file der
Aufgabenstellung: Protokoll von Johnnie Akgün 0401350 Identifizierung von strukturierten non coding RNAs von 6 verschiedenen Aspergillus Stämmen. Arbeitsschritte: Begonnen wird mit einem Fasta file der
Bayesianische Netzwerke - Lernen und Inferenz
 Bayesianische Netzwerke - Lernen und Inferenz Manuela Hummel 9. Mai 2003 Gliederung 1. Allgemeines 2. Bayesianische Netzwerke zur Auswertung von Genexpressionsdaten 3. Automatische Modellselektion 4. Beispiel
Bayesianische Netzwerke - Lernen und Inferenz Manuela Hummel 9. Mai 2003 Gliederung 1. Allgemeines 2. Bayesianische Netzwerke zur Auswertung von Genexpressionsdaten 3. Automatische Modellselektion 4. Beispiel
Bioinformatik. Multiple Sequence Alignment Sum-of-pairs Score. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Ulf Leser Wissensmanagement in der Bioinformatik Quasar Grundidee Search-Phase sucht Regionen mit Länge w und hoher Ähnlichkeit D.h. Regionen
Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Ulf Leser Wissensmanagement in der Bioinformatik Quasar Grundidee Search-Phase sucht Regionen mit Länge w und hoher Ähnlichkeit D.h. Regionen
Effiziente Algorithmen und Komplexitätstheorie
 Fakultät für Informatik Lehrstuhl 2 Vorlesung Effiziente Algorithmen und Komplexitätstheorie Sommersemester 2008 Ingo Wegener Ingo Wegener 03. Juli 2008 1 Sequenzanalyse Hauptproblem der Bioinformatik
Fakultät für Informatik Lehrstuhl 2 Vorlesung Effiziente Algorithmen und Komplexitätstheorie Sommersemester 2008 Ingo Wegener Ingo Wegener 03. Juli 2008 1 Sequenzanalyse Hauptproblem der Bioinformatik
Informationsgehalt von DNA
 Informationsgehalt von DNA Topics Genes code, gene organisation, signals, gene detection Genomes genome organisation, nucleotide patterns, junk DNA DNA als Informationsträger DNA Building Blocks Desoxyribose
Informationsgehalt von DNA Topics Genes code, gene organisation, signals, gene detection Genomes genome organisation, nucleotide patterns, junk DNA DNA als Informationsträger DNA Building Blocks Desoxyribose
Mathematik in den Life Siences
 Gerhard Keller Mathematik in den Life Siences Grundlagen der Modellbildung und Statistik mit einer Einführung in die Statistik-Software R 49 Abbildungen Verlag Eugen Ulmer Stuttgart Inhaltsverzeichnis
Gerhard Keller Mathematik in den Life Siences Grundlagen der Modellbildung und Statistik mit einer Einführung in die Statistik-Software R 49 Abbildungen Verlag Eugen Ulmer Stuttgart Inhaltsverzeichnis
Einführung in die Angewandte Bioinformatik: Algorithmen und Komplexität; Multiples Alignment und
 Einführung in die Angewandte Bioinformatik: Algorithmen und Komplexität; Multiples Alignment 04.06.2009 und 18.06.2009 Prof. Dr. Sven Rahmann 1 Zwischenspiel: Algorithmik Bisher nebenbei : Vorstellung
Einführung in die Angewandte Bioinformatik: Algorithmen und Komplexität; Multiples Alignment 04.06.2009 und 18.06.2009 Prof. Dr. Sven Rahmann 1 Zwischenspiel: Algorithmik Bisher nebenbei : Vorstellung
Stream Processing II
 Stream Processing II K-Buckets Histogram Histogramme sind graphische Darstellungen der Verteilung von numerischen Werten Werden durch Intervalle, die sich nicht überlappen, dargestellt Ein Intervall wird
Stream Processing II K-Buckets Histogram Histogramme sind graphische Darstellungen der Verteilung von numerischen Werten Werden durch Intervalle, die sich nicht überlappen, dargestellt Ein Intervall wird
Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR)
") PCR Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR) von Kary B. Mullis entwickelt (1985) eigentlich im Mai 1983 bei einer nächtlichen Autofahrt erstes erfolgreiches Experiment am 16.12.1983
PCR Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR) von Kary B. Mullis entwickelt (1985) eigentlich im Mai 1983 bei einer nächtlichen Autofahrt erstes erfolgreiches Experiment am 16.12.1983
Bioinformatik. Alignment mit linearem Platzbedarf K-Band Alignment. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Alignment mit linearem Platzbedarf K-Band Alignment Ulf Leser Wissensmanagement in der Bioinformatik Ankündigungen Gastvortrag: Morgen Dr. Klein, Metanomics GmbH Bioinformatik in der Pflanzengenomik
Bioinformatik Alignment mit linearem Platzbedarf K-Band Alignment Ulf Leser Wissensmanagement in der Bioinformatik Ankündigungen Gastvortrag: Morgen Dr. Klein, Metanomics GmbH Bioinformatik in der Pflanzengenomik
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
AlgoBio WS 16/17 Differenzielle Genexpression. Annalisa Marsico
 AlgoBio WS 16/17 Differenzielle Genexpression Annalisa Marsico 04.01.2017 Pipeline für die Mikroarray-Analyse Bildanalyse Hintergrundkorrektur Normalisierung Vorverarbeitung Zusammenfassung Quantifizierung
AlgoBio WS 16/17 Differenzielle Genexpression Annalisa Marsico 04.01.2017 Pipeline für die Mikroarray-Analyse Bildanalyse Hintergrundkorrektur Normalisierung Vorverarbeitung Zusammenfassung Quantifizierung
Welche Alignmentmethoden haben Sie bisher kennengelernt?
 Welche Alignmentmethoden haben Sie bisher kennengelernt? Was heißt optimal? Optimal = die wenigsten Mutationen. Sequenzen bestehen aus Elementen (z.b. Aminosäuren oder Nukleotide). Edit Distanzen sind
Welche Alignmentmethoden haben Sie bisher kennengelernt? Was heißt optimal? Optimal = die wenigsten Mutationen. Sequenzen bestehen aus Elementen (z.b. Aminosäuren oder Nukleotide). Edit Distanzen sind
Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken
 Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken 14.05.2009 Prof. Dr. Sven Rahmann 1 3 Proteinsequenz-Datenbanksysteme NCBI Entrez Proteins EBI SRS Proteins UniProt (empfohlen) 2
Einführung in die Angewandte Bioinformatik: Proteinsequenz-Datenbanken 14.05.2009 Prof. Dr. Sven Rahmann 1 3 Proteinsequenz-Datenbanksysteme NCBI Entrez Proteins EBI SRS Proteins UniProt (empfohlen) 2
FOR MUW SSM3 (2008) STUDENTS EDUCATIONAL PURPOSE ONLY
 STUDENTS EDUCATIONAL PURPOSE ONLY") Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
Signifikanz von Alignment Scores und BLAST
 Westfälische Wilhelms Universität Münster Fachbereich 10 - Mathematik und Informatik Signifikanz von Alignment Scores und BLAST Seminarvortrag von Leonie Zeune 10. Mai 2012 Veranstaltung: Seminar zur mathematischen
Westfälische Wilhelms Universität Münster Fachbereich 10 - Mathematik und Informatik Signifikanz von Alignment Scores und BLAST Seminarvortrag von Leonie Zeune 10. Mai 2012 Veranstaltung: Seminar zur mathematischen
BLAST Basic Local Alignment Search Tool
 BLAST Basic Local Alignment Search Tool Martin Winkels 21.12.2012 wissen leben WWU Münster Institut für Medizinische Informatik Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis 1 Einleitung 2 2
BLAST Basic Local Alignment Search Tool Martin Winkels 21.12.2012 wissen leben WWU Münster Institut für Medizinische Informatik Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis 1 Einleitung 2 2
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Center-Star Score Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Multiples Sequenzalignment Sum-Of-Pair
Algorithmische Bioinformatik Multiple Sequence Alignment Sum-of-pairs Score Center-Star Score Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Multiples Sequenzalignment Sum-Of-Pair
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de") Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
a i j (B + C ) j k = n (a i j b j k + a i j b j k ) =
 j k = n (a i j b j k + a i j b j k ) =") Lösungen Lineare Algebra für Physiker, Serie 2 Abgabe am 25.10.2007 1. Es seien A K m n, B,C K n p und D K p q gegeben. 9 P (a) Beweisen Sie das Distributivgesetz A(B + C ) = A B + AC. (b) Beweisen Sie
Lösungen Lineare Algebra für Physiker, Serie 2 Abgabe am 25.10.2007 1. Es seien A K m n, B,C K n p und D K p q gegeben. 9 P (a) Beweisen Sie das Distributivgesetz A(B + C ) = A B + AC. (b) Beweisen Sie
Lineare Algebra - Übungen 1 WS 2017/18
 Prof. Dr. A. Maas Institut für Physik N A W I G R A Z Lineare Algebra - Übungen 1 WS 017/18 Aufgabe P1: Vektoren Präsenzaufgaben 19. Oktober 017 a) Zeichnen Sie die folgenden Vektoren: (0,0) T, (1,0) T,
Prof. Dr. A. Maas Institut für Physik N A W I G R A Z Lineare Algebra - Übungen 1 WS 017/18 Aufgabe P1: Vektoren Präsenzaufgaben 19. Oktober 017 a) Zeichnen Sie die folgenden Vektoren: (0,0) T, (1,0) T,
Technische Universität München Zentrum Mathematik. Übungsblatt 12
 Technische Universität München Zentrum Mathematik Mathematik 1 (Elektrotechnik) Prof. Dr. Anusch Taraz Dr. Michael Ritter Übungsblatt 12 Hausaufgaben Aufgabe 12.1 Sei f : R 3 R 3 gegeben durch f(x) :=
Technische Universität München Zentrum Mathematik Mathematik 1 (Elektrotechnik) Prof. Dr. Anusch Taraz Dr. Michael Ritter Übungsblatt 12 Hausaufgaben Aufgabe 12.1 Sei f : R 3 R 3 gegeben durch f(x) :=
MBI: Sequenzvergleich ohne Alignment
 MBI: Sequenzvergleich ohne Alignment Bernhard Haubold 12. November 2013 Wiederholung Exaktes & inexaktes Matching Das exakte Matching Problem Naive Lösung Präprozessierung Muster(Pattern): Z-Algorithmus,
MBI: Sequenzvergleich ohne Alignment Bernhard Haubold 12. November 2013 Wiederholung Exaktes & inexaktes Matching Das exakte Matching Problem Naive Lösung Präprozessierung Muster(Pattern): Z-Algorithmus,
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Ein progressiver Algorithmus für Multiple Sequence Alignment auf Basis von A*-Suche. Christine Zarges
 Ein progressiver Algorithmus für Multiple Sequence Alignment auf Basis von A*-Suche Christine Zarges Algorithm Engineering Report TR07-1-009 November 2007 ISSN 1864-4503 Fakultät für Informatik Algorithm
Ein progressiver Algorithmus für Multiple Sequence Alignment auf Basis von A*-Suche Christine Zarges Algorithm Engineering Report TR07-1-009 November 2007 ISSN 1864-4503 Fakultät für Informatik Algorithm
Serie 5. Lineare Algebra D-MATH, HS Prof. Richard Pink. 1. [Aufgabe] Invertieren Sie folgende Matrizen über Q:
![Serie 5. Lineare Algebra D-MATH, HS Prof. Richard Pink. 1. [Aufgabe] Invertieren Sie folgende Matrizen über Q:](/thumbs/72/66491799.jpg "Serie 5. Lineare Algebra D-MATH, HS Prof. Richard Pink. 1. [Aufgabe] Invertieren Sie folgende Matrizen über Q:") Lineare Algebra D-MATH, HS 214 Prof Richard Pink Serie 5 1 [Aufgabe] Invertieren Sie folgende Matrizen über Q: 1 a) 1 1 1 1 1 2 1 1 1 b) 1 2 1 1 1 1 2 1 1 1 1 2 1 2 3 1 c) 1 3 3 2 2 1 5 3 1 2 6 1 [Lösung]
Lineare Algebra D-MATH, HS 214 Prof Richard Pink Serie 5 1 [Aufgabe] Invertieren Sie folgende Matrizen über Q: 1 a) 1 1 1 1 1 2 1 1 1 b) 1 2 1 1 1 1 2 1 1 1 1 2 1 2 3 1 c) 1 3 3 2 2 1 5 3 1 2 6 1 [Lösung]
Genannotation bei Prokaryoten
 Genannotation bei Prokaryoten Maike Tech Abt. Bioinformatik Institut für Mikrobiologie und Genetik (IMG) Universität Göttingen 28. November 2005 Genetik von Pro- und Eukaryoten Eukaryoten Prokaryoten Zellkern
Genannotation bei Prokaryoten Maike Tech Abt. Bioinformatik Institut für Mikrobiologie und Genetik (IMG) Universität Göttingen 28. November 2005 Genetik von Pro- und Eukaryoten Eukaryoten Prokaryoten Zellkern
Sequenzen-Alignierung in der Bioinformatik
 Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 22. VO 23..26 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 22. VO 23..26 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Bioinformatik an der FH Bingen
 Bioinformatik an der FH Bingen Prof. Dr. Antje Krause 05.11.2010 Wie alles begann... 1955 erste Proteinsequenz (nach 12 Jahren Arbeit) veröffentlicht (Insulin vom Rind) Frederick Sanger MALWTRLRPLLALLALWPPPPA
Bioinformatik an der FH Bingen Prof. Dr. Antje Krause 05.11.2010 Wie alles begann... 1955 erste Proteinsequenz (nach 12 Jahren Arbeit) veröffentlicht (Insulin vom Rind) Frederick Sanger MALWTRLRPLLALLALWPPPPA
Einführung in die Angewandte Bioinformatik: BLAST E-Werte, Algorithmen, Multiple Alignments, R
 Einführung in die Angewandte Bioinformatik: BLAST E-Werte, Algorithmen, Multiple Alignments, R 17.06.2010 Prof. Dr. Sven Rahmann 1 Protein BLAST (blastp) Hier wird die Sequenz eingegeben oder hochgeladen,
Einführung in die Angewandte Bioinformatik: BLAST E-Werte, Algorithmen, Multiple Alignments, R 17.06.2010 Prof. Dr. Sven Rahmann 1 Protein BLAST (blastp) Hier wird die Sequenz eingegeben oder hochgeladen,
Lokale Sequenzähnlichkeit. Genomische Datenanalyse 9. Kapitel
 Lokale Sequenzähnlichkeit Genomische Datenanalyse 9. Kapitel Globale Sequenzähnlichkeit: Zwei Cytochrome C Sequenzen: Eine vom Menschen und eine aus der Maus. Die Sequenzen sind gleich lang, man kann sie
Lokale Sequenzähnlichkeit Genomische Datenanalyse 9. Kapitel Globale Sequenzähnlichkeit: Zwei Cytochrome C Sequenzen: Eine vom Menschen und eine aus der Maus. Die Sequenzen sind gleich lang, man kann sie
Algorithmen zum Strukturvergleich Strukturelle Bioinformatik WS16/17
 Algorithmen zum Strukturvergleich Strukturelle Bioinformatik WS16/17 Dr. Stefan Simm, 01.11.2016 simm@bio.uni-frankfurt.de Strukturvergleich, -alignment und -superposition SPEZIFIKATION DES PROBLEMS Detailgrad
Algorithmen zum Strukturvergleich Strukturelle Bioinformatik WS16/17 Dr. Stefan Simm, 01.11.2016 simm@bio.uni-frankfurt.de Strukturvergleich, -alignment und -superposition SPEZIFIKATION DES PROBLEMS Detailgrad