Medizintechnik: Regulatorische Rahmenbedingungen I
|
|
|
- Willi Fiedler
- vor 8 Jahren
- Abrufe
Transkript
1 Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Reinhard Busse, Prof. Dr. med. MPH FFPH FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating Centre for Health Systems Research and Management) & European Observatory on Health Systems and Policies 1
 & European")
2 Datum Inhalt der Lehrveranstaltung Dozent/in Einführungsveranstaltung Busse Medizintechnik-Industrie Marktentwicklung Busse Regulatorische Rahmenbedingungen I Busse Regulatorische Rahmenbedingungen II Busse Kundenmanagement Busse Telemedizin und e-health Henschke Pharmazeutische Industrie Marktentwicklung Busse Regulatorische Rahmenbedingungen I Busse Regulatorische Rahmenbedingungen II Henschke Preisbildung Busse Evaluation und Pharmakoökonomie Busse Kundenmanagement Busse Klausur Henschke 2

3 Definition: Medizinprodukte ( 3 MPG) Medizinprodukte sind alle einzeln oder miteinander verbunden verwendeten Instrumente, Apparate, Vorrichtungen [ ], die vom Hersteller zur Anwendung für Menschen mittels ihrer Funktionen zum Zwecke der a) der Erkennung, Verhütung, Überwachung, Behandlung oder Linderung von Krankheiten b) der Erkennung, Überwachung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen, c) der Untersuchung, der Ersetzung oder der Veränderung des anatomischen Aufbaus oder eines physiologischen Vorgangs oder d) der Empfängnisregelung zu dienen bestimmt sind [ ] und die im Gegensatz zu Arzneimitteln ihre Hauptwirkung nicht auf pharmakologischem, immunologischem oder metabolischem Wege hervorbringen. 3
 der Empfängnisregelung zu dienen bestimmt sind [ ] und die im Gegensatz zu Arzneimitteln ihre Hauptwirkung nicht auf pharmakologischem, immunologischem oder metabolischem Wege")
4 Inverkehrbringung von Medizinprodukten I Voraussetzung für das Inverkehrbringen (Marktzulassung): CE-Kennzeichnung Anbringung der CE-Kennzeichnung erfordert: - Erfüllung der grundlegenden Anforderungen durch Anwendung von Normen - Durchführung vorgeschriebener Konformitätsbewertungsverfahren verbindliche RL für MP beschränken sich auf die Nennung sog. grundlegender Anforderungen der zutreffenden RL: -90/385/EWG (aktive implantierbare Medizinprodukte) -98/79/EWG (In-vitro-Diagnostika) - 93/42/EWG (sonstige Medizinprodukte) detaillierte technische Spezifikationen sind durch privatwirtschaftliche Normenorganisationen ausgearbeitet (auf diese Normen wird im Gesetz verwiesen) Vorteil: Technische Ausführungsanforderungen sind schneller an den technischen Fortschritt anpassbar 4
 Vorteil: Technische Ausführungsanforderungen sind")
5 Inverkehrbringung von Medizinprodukten I Voraussetzung für das Inverkehrbringen (Marktzulassung): CE-Kennzeichnung Ein Aktives Medizinprodukt ist ein Medizinprodukt, dessen Betrieb von einer Stromquelle oder einer anderen Energiequelle Anbringung der CE-Kennzeichnung erfordert: (mit Ausnahme der direkt vom menschlichen - Erfüllung Körper oder der durch grundlegenden die Schwerkraft erzeugten Anforderungen durch Anwendung von Normen - Durchführung Energie) vorgeschriebener abhängig ist. Konformitätsbewertungsverfahren In-vitro-Diagnostikum(IVD) ist jedes Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibriermaterial, Kontrollmaterial, Kit, Instrument, Apparat, Gerät oder System einzeln oder in Verbindung miteinander nach der vom Hersteller festgelegten Zweckbestimmung zur In-vitro-Untersuchung von aus dem Körper stammenden Proben, einschließlich Blutund Gewebespenden, verwendet wird und ausschließlich oder hauptsächlich dazu dient, Informationen zu liefern: -über physiologische oder pathologische Zustände verbindliche RL für MP beschränken sich auf die Nennung sog. grundlegender oder Anforderungen der zutreffenden RL: -90/385/EWG (aktive implantierbare Medizinprodukte) -98/79/EWG (In-vitro-Diagnostika) - 93/42/EWG (sonstige Medizinprodukte) - über angeborene Anomalien oder -zur Prüfung auf Unbedenklichkeit und Verträglichkeit bei den potentiellen Empfängern oder -zur Überwachung therapeutischer Maßnahmen. CAVE: In-vivo-Diagnostika (z.b. Kontrastmittel) fallen in Deutschland unter das Arzneimittelgesetz! detaillierte technische Spezifikationen sind durch privatwirtschaftliche Normenorganisationen ausgearbeitet (auf diese Normen wird im Gesetz verwiesen) Vorteil: Technische Ausführungsanforderungen sind schneller an den technischen Fortschritt anpassbar 5
 ist jedes Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibriermaterial, Kontrollmaterial, Kit, Instrument, Apparat, Gerät oder System")
6 Inverkehrbringung von Medizinprodukten II Art des Konformitätsbewertungsverfahren sowie den Umfang der Beteiligung einer unabhängigen Prüf- und Zertifizierungsstelle (Benannte Stelle) ist vom potenziellen (Gesundheits-)Risiko der Produkte nicht von ihrer Wirksamkeit abhängig (Logik des Binnenmarktes!) aktive implantierbare Medizinprodukte (90/385/EWG): keine weitere Unterscheidung nach Risikogesichtspunkten MP nach RL 93/42/EWG: Produktdifferenzierung nach festgelegten Kriterien in Klassen In-vitro-Diagnostika (98/79/EG): Einteilung in Gruppen Konformitätsbewertungsverfahren sowie deren Durchführung sind in der dt. Verordnung über Medizinprodukte (MPV) geregelt. Diese verweist auf entsprechende Anhänge der europäischen Richtlinien. 6
: Einteilung in Gruppen Konformitätsbewertungsverfahren sowie deren Durchführung sind in der dt. Verordnung über Medizinprodukte (MPV) geregelt.")
7 Marktzulassung von Medizinprodukten einheitliches Zulassungsverfahren in Europa wird jeweils von Benannten Stellen (Zertifizierungsstellen) durchgeführt in Deutschland derzeit 15, z. B. TÜV, Materialprüfstellen in der EU insgesamt ca. 80 Benennende Behörden der Benannten Stellen in Dtl: Benennung und Überwachung der Zertifizierungsstellen ZLG-Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten: nichtaktiven Medizinprodukte und In-vitro-Diagnostika ZLS- Zentralstelle der Länder für Sicherheitstechnik: aktive Medizinprodukte Ziel: Risiko für den Verbraucher bzw. den Patienten zu minimieren 3 Hersteller muss Produktsicherheit und medizinisch-technische Leistungsfähigkeit nachweisen Zulassung basiert auf Überprüfung der Qualität und der Qualitätssicherungsmaßnahmen 7

8 Ausschnitt aus der Liste der Benannten Stellen 8

9 Infos zu Benannten Stellen für Medizinprodukte 9


10 Ausschnitte des Geltungsbereiches Berlin Cert 10

11 Klassifizierung von Medizinprodukten nach Richtlinie 93/42/EWG Wozu? zur Abschätzung des potentiellen Risikos Wie? durch die Einteilung in Risikoklassen (I, IIa, IIb, III) Wonach? Anwendung der 18 Regeln im Anhang IX der Richtlinie (93/42/EWG) unter Berücksichtigung aller Charakteristika des MP Zweckbestimmung Angewandte Technologie (aktiv, nicht aktiv...) Anwendungsart, -dauer (<1h, <30d, >30d), -ort 11
 unter Berücksichtigung aller")
12 Klassen von Medizinprodukten nach Richtlinie 93/42/EWG Einteilung der Medizinprodukte nach Art in Klassen: I Produkte mit niedrigem Risiko, die meisten nicht-invasiven Produkte und wiederverwendbare chirurgische Instrumente (z.b. Stethoskope, Spatel) IIa nicht aktive Produkte mit mittlerem Risiko, invasive und nicht- invasive Produkte für kurzzeitige Benutzung (z.b. Kanülen) IIb aktive Produkte mit mittlerem Risiko, die Substanzen oder Energie mit potentiellem Risiko emittieren (z.b. Röntgengeräte), und Produkte für längere Nutzung III Produkte mit hohem Risiko undsolche, die mit dem Gefäßsystem oder dem zentralen Nervensystem in Kontakt kommen (z.b. Gefäßtransplantate) Zunehmendes Gefährdungspotenzial des Medizinproduktes 12
 Zunehmendes Gefährdungspotenzial des Medizinproduktes 12")
13 Klassifizierungsregeln Anhang IX der RL 93/42/EWG Klassifizierungsregeln Nicht invasive Invasive Zusätzliche Regeln für Besondere Produkte Produkte aktive Produkte Regeln Regel 1-4 Regel 5-8 Regel 9-12 Regel

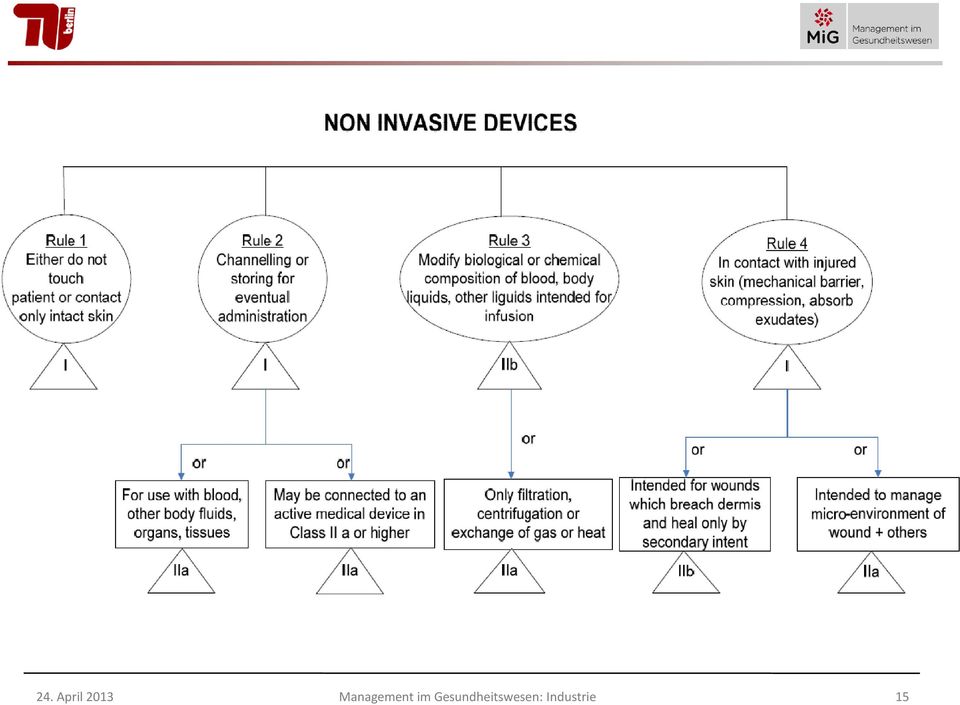
14 Wesentliche Klassifizierungsmerkmale von nicht-invasiven Medizinprodukten Regel 1: alle nicht invasiven Produkte, für die keine andere Regel zutrifft: Klasse I Regel 2: Produkte zur Durchleitung von Blut oder zur Aufbewahrung von Blut etc., oder Einsatz mit Produkten der Klasse IIa oder höher: Klasse IIa Regel 3: Produkte zur Veränderung der biologischen und chemischen Zusammensetzung des Blutes, wenn in den Körper perfundiert: Klasse IIb Regel 4: Produkte, die in Berührung mit verletzter Haut kommen als mechanische Barriere: Klasse IIa - mit Beeinflussung der Mikroumgebung einer Wunde: Klasse IIa - bei Durchtrennung der Dermis: Klasse IIb 14

15 15
16 Wesentliche Klassifizierungsmerkmale von invasiven Medizinprodukten I Regel 5: Produkte (außer chirurgisch-invasiven) für - vorübergehende Anwendung: Klasse I - kurzzeitige Anwendung und bei Verwendung zusammen mit aktiven Produkten der Klasse IIa oder höher: Klasse IIb Regel 6: - Wiederverwendbare chirurgische Instrumente: Klasse I - chirurgisch-invasive Produkte für die vorübergehende Anwendung: Klasse IIa - Produkte zur Energieabgabe (ionisierende Strahlung): Klasse IIb - Produkte mit biologischer Wirkung: Klasse IIb - Produkte zur Verabreichung von Arzneimitteln mit potentieller Gefährdung: Klasse IIb - Produkte, die resorbiert werden: Klasse IIb - Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III 16

17 Wesentliche Klassifizierungsmerkmale von invasiven Medizinprodukten II Regel 7: - Chirurgisch-invasive Produkte für kurzzeitige Anwendung: Klasse IIa - Produkte zur Energieabgabe (ionisierende Strahlung) und zur Abgabe von Arzneimitteln: Klasse IIb - Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III - Produkte in Kontakt mit dem zentralen Nervensystem: Klasse III - Produkte mit biologischer Wirkung oder Resorption: Klasse III Regel 8: - Produkte zur Implantierung in Zähne: Klasse IIa - alle implantierbaren Produkte und chirurgisch-invasiven Produkte zur langzeitigen Anwendung, wenn keine andere Regel zutrifft: Klasse IIb - Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III - Produkte in Kontakt mit dem zentralen Nervensystem: Klasse III - Produkte mit biologischer Wirkung oder Resorption: Klasse III - Produkte zur Abgabe von Arzneimitteln: Klasse III 17

18 Zusatzregeln für aktive(energiebetriebene) Produkte Regel 9: - alle therapeutischen Produkte zur Abgabe oder zum Austausch von Energie: Klasse IIa - alle therapeutischen Produkte zur Abgabe oder zum Austausch von Energie bei potentieller Gefährdung: Klasse IIb - alle aktiven Produkte zur Leistungssteuerung oder Leistungskontrolle von aktiven therapeutischen Produkten: Klasse IIb Regel 10: - alle aktiven diagnostischen Produkte zur Abgabe von Energie: Klasse IIa - Produkte zur In-vivo-Darstellung von Radiopharmaka: Klasse IIa - alle aktiven diagnostischen Produkte zur Abgabe von Energie mit potentieller Gefährdung: Klasse Iib - Produkte zur Aussendung ionisierender Strahlung und Produkte zur Steuerung oder Kontrolle solcher Produkte: Klasse IIb Regel 11: - alle aktiven Produkte zur Abgabe von Arzneimitteln, Körperflüssigkeiten und anderen Stoffen an den Körper oder/und für deren Entfernung: Klasse IIa - alle aktiven Produkte zur Abgabe von Arzneimitteln, Körperflüssigkeiten oder/und für deren Entfernung bei potentieller Gefährdung: Klasse IIb Regel 12: alle anderen aktiven Produkte: Klasse I 18

19 Besondere Regeln Regel 13: Produkte zu deren Bestandteil ein Stoff gehört, der bei gesonderter Verwendung als Arzneimittel angesehen werden kann: Klasse III Regel 14: - Produkte zur Empfängnisverhütung und zum Schutz vor der Übertragung sexuell übertragbaren Krankheiten: IIb - Produkte zur Empfängnisverhütung und zum Schutz vor der Übertragung sexuell übertragbaren Krankheiten,wenn diese implantiert werden oder wenn es sich um invasive Produkte zur langzeitigen Anwendung handelt: Klasse III Regel 15: - Produkte speziell zur Desinfektion von Produkten: Klasse IIa (neu: Desinfektionsmittel für invasive MP: IIb) - Produkte zur Desinfektion, Reinigung, Spülung und zum Hydratisieren von Kontaktlinsen: Klasse IIb Regel 16: Nicht-aktive Produkte zur Aufzeichnung von Röntgendiagnosebildern: Klasse IIa (neu: digitaler Röntgenbildempfänger: IIb) Regel 17: Produkte, die unter Verwendung von tierischen Geweben oder Folgeerzeugnissen hergestellt werden: Klasse III Regel 18: Blutbeutel: Klasse IIb 24. April Management im Gesundheitswesen: Industrie
 - Produkte zur Desinfektion, Reinigung, Spülung und zum Hydratisieren von Kontaktlinsen: Klasse IIb Regel 16: Nicht-aktive Produkte zur")
20 Klassifizierung eines Medizinproduktes Beispiel Gehhilfe 1. Welche Richtlinie gilt? Ausschlussverfahren - Eine Gehhilfe ist kein aktives implantierbares Medizinprodukt (keine Energiequelle, keine Implantation) 90/385/EWG ist nicht anwendbar - Eine Gehhilfe ist kein in vitro Diagnostikum 98/79/EG ist nicht anwendbar - Nach dem Ausschlussverfahren ist die Richtlinie 93/42/EWG anzuwenden 2. Klassifizierung? Überprüfen der Klassifizierungsregeln - Gemäß den Klassifizierungsregeln des Anhangs IX der Richtlinie 93/42/EWG ist eine Gehhilfe ein Produkt der Klasse I, denn - Regel 1: alle nicht invasiven Produkte gehören der Klasse I an, es sei denn, es findet eine der folgenden Regeln Anwendung - Regel 2: nicht invasives MP dient der Durchleitung oder Aufbewahrung von Körperflüssigkeiten... IIa trifft nicht zu - Regel 3: nicht invasives MP zur Veränderung der biologischen oder chemischen Zusammensetzung des Blutes... IIa trifft nicht zu - Regel 4: Nicht invasive MP die mit verletzter Haut in Berührung kommen... IIa, IIb trifft nicht zu 20

21 Einige konkrete Beispiele für Medizinprodukte: Klasse I Klasse IIa Klasse IIb Klasse III ärztliche Instrumente Gehhilfen Rollstühle Spitalbetten Stützstrümpfe Verbandmittel Wiederverwendbare chirurgische Instrumente Dentalmaterialien diagnostische Ultraschallgeräte Einmalspritzen Hörgeräte Kontaktlinsen Trachealtuben Zahnkronen Anästhesiegeräte Beatmungsgeräte Bestrahlungsgeräte Blutbeutel Defibrillatoren Dialysegeräte Kondome Kontaktlinsenreiniger Dentalimplantate Herzkatheter künstliche Gelenke Stents resorbierbares chirurgisches Nahtmaterial Intrauterinpessar (Spirale) Brustimplantat 21
22 Zulassungsregulation der Produktklassen nach RL 93/42/EWG Klassifizierung der Medizinprodukte gem. Anhang IX Medizinprodukte-RL Klassen I, IIa, IIb, III I, IIa EG-Konformitätserklärung gemäß Anhang VII IIb, III EG-Baumusterprüfung gemäß Anhang III IIa, IIb, III EG-Konformitätserklärung gemäß Anhang II vollständiges Qualitätssicherungssystem I I 1, IIa, IIb, III EG-Prüfung gemäß Anhang IV I 1, IIa, IIb, III EG-Konformitätserklärung gemäß Anhang V (Qualitätssicherung der Produktion) I 1, IIa, IIb EG-Konformitätserklärung gemäß Anhang VI (Qualitätssicherung der Produkte) IIa, IIb ohne Prüfung der Produktauslegung (Abschnitt 4) III mit Prüfung der Produktauslegung (Abschnitt 4) CE Beteiligung einer benannten Stelle nicht erforderlich CE + Kennnummer der benannten Stelle Beteiligung einer benannten Stelle erforderlich Umfang der Beteiligung ist abhängig vom Risikograd des Medizinproduktes 1 Produkte der Klasse I, die steril in den Verkehr gebracht werden, oder Produkte der Klasse I mit Messfunktion 22
23 Beispiele von Zertifikaten 23
24 Beispiele von Zertifikaten 24
25 RL 2007/47/EG zur Änderung der RL 90/385/EWG u. 93/42/EWG - Aufnahme von Software in die Definition MP - Verschärfung der Konformitätsbewertung in den Anhängen der RL - z.b. Benannte Stelle muss Stichprobe für jede generische Produktgruppe bei MP der Klasse IIa und IIb prüfen - Längere Aufbewahrungsfristen für Hersteller-Unterlagen - Änderung der Klassifizierungskriterien: - Eigenständige Software aktives MP - Konzept der ununterbrochenen Anwendung - Regel 15: Desinfektionsmittel für invasive MP Klasse IIb - Konsultationsverfahren für MP mit unterstützendem Arzneistoffanteil - Anpassung der RL 90/385/EWG an die RL 93/42/EWG 25
26 Gesetz zur Änderung medizinproduktrechtlicher Vorschriften Ziel: - Umsetzung der RL 2007/47/EG Inhalte: - Klinische Prüfung bzw. Leistungsbewertungsprüfungen: - Zentrale Genehmigung durch BfArM/PEI (statt Anzeige bei LB) und - positive Zustimmung einer nach Landesrecht gebildeten Ethik-Kommission - Einführung von - Meldepflicht bei allen schwerwiegenden unerwünschten Ereignissen in klinischen Studien (SAE s seriousadverseevents) von Prüfer und Sponsor (Doppelmeldung) - entsprechenden Bewertungsverfahren im Rahmen von klinischen Prüfungen 26
27 Klare Aufgabentrennung Gesetz zur Änderung medizinproduktrechtlicher Vorschriften - Zentrale Genehmigungsbehörden (BfArM und PEI) prüfen nach wissenschaftlichen und technischen Gesichtspunkten - Technische Sicherheit - Biokompatibilität - Wissenschaftlichkeit des Prüfplans - Generelle Nutzen-/ Risikobewertung Genehmigungs- statt Anzeigepflicht - Landesrechtlich eingerichteten Ethik-Kommission prüft nach ethischen und rechtlichen Gesichtspunkten - die Zulässigkeit der klinischen Prüfung 24. April Management im Gesundheitswesen: Industrie
28 Gesetz zur Änderung medizinproduktrechtlicher Vorschriften Zweck einer klinische Prüfung: - Erbringung des Nachweises, dass Leistungen des MP bei normalen Einsatzbedingungen den grundlegenden Anforderungen entsprechen (CAVE: Unterschied zu [klinischer] Wirksamkeit) - Zweckbestimmung muss sehr genau beschrieben werden (Funktion, Indikation, Kontraindikation) - Ermittlung unerwünschter Nebenwirkungen - Sicherheitsregelungen zur Durchführung von klinischen Prüfungen verlangen, dass Produkte alle sicherheitstechnischen Eigenschaften aufweisen 28
29 Gesetz zur Änderung medizinproduktrechtlicher Vorschriften Der Hersteller hat für jedes MP u. a. den Nachweis zu erbringen, dass: - die Anwendung des Medizinproduktes bei normalen Einsatzbedingungen den klinischen Zustand des Patienten die Sicherheit des Patienten die Sicherheit und Gesundheit des Anwenders und Dritter nicht gefährdet -die angepriesene Leistungsfähigkeit des Produktes unter normalen Bedingungen erreicht wird. Dieser Nachweis kann durch eine Zusammenstellung der derzeit verfügbaren Literatur erbracht werden. Für Hochrisikoprodukte der Klasse IIbund III sind klinische Prüfungen erforderlich. 24. April 2013 Management im Gesundheitswesen: Industrie 29
30 Ablauf: Entwicklung eines Medizinproduktes Medizinprodukt Technische Entwicklung Produktion Biokompatibilität Technische Leistung Anwendung an Patienten (partiell an Probanden) Zertifizierung Arzneimittel Technische Entwicklung Produktion Toxologie Pharmakokinetik Pharmakodynamik Anwendung an Probanden/ Patienten (Phase I-III) Zulassung Langzeitbeobachtung Post Market Clinical Follow-up Marktsüberwachung Phase IV Schorn GH (2010): Klinische Prüfungen von Medizinprodukten: Fragen und Antworten zum geänderten MP-Recht. In: Medizinprodukte Journal 17 (3)
31 Zertifizierung bedeutet nicht automatisch Kostenübernahme durch die GKV ( Reg. Rahmenbed. II) 31
32 Erweiterung des üblichen Dreiecks bei Medizinprodukten (I) Zahler (GKV) Beitrag/Steuern Vergütung für Produkt und/oder Dienstleistung Regulierer Patient med. Leistungserbringer (z.b. Krankenhaus) Zuzahlungen Hilfsmitteldistributeur (z.b. Sanitätshaus) Hersteller 32
33 Strukturierung von Medizinprodukten unter Vergütungsgesichtspunkten Medizinprodukte Kategorie I Hilfsmittel i.d.r. Standardprodukte, die einem individuellen Patienten verschrieben und gegeben werden (z.b. Inkontinenzunterlagen, Rollstühle) Produkt, nicht Dienstleistung steht im Mittelpunkt Kategorie II Implantate & Endo- Prothesen Medizinprodukte, die einem individuellen Patienten implantiert oder angepasst werden (z.b. Gelenk- Endoprothesen, Stents, Herzschrittmacher) zumeist steht die Dienstleistung im Vordergrund Kategorie III Medizintechnik zur Arzt-Unterstützung TechnischeGeräte, die Ärzte bei Diagnostikund Behandlung unterstützen (Röntgen, Endoskope); Finanzierung in zwei Stufen: IIIa: Investition IIIb: Refinanzierung über Dienstleistungen 33
34 Erweiterung des üblichen Dreiecks bei Medizinprodukten (II) Zahler (GKV) Beitrag/Steuern Vergütung für Produkt und/oder Dienstleistung II III b Patient II III b med. Leistungserbringer (z.b. Krankenhaus) Zuzahlungen I (Investition) III a I II Hilfsmitteldistributeur (z.b. Sanitätshaus) I Hersteller NB: I, II, IIIa und IIIb beziehen sich auf die Kategorien in der vorherigen Abbildung 34
Klassifizierung von Medizinprodukten
 Klassifizierung von Medizinprodukten Die Medizinprodukte-Richtlinie 93/42/EWG sieht vor, dass jedes Medizinprodukt einer bestimmten Klasse zugeordnet werden muss. Von dieser Zuordnung hängt das weitere
Klassifizierung von Medizinprodukten Die Medizinprodukte-Richtlinie 93/42/EWG sieht vor, dass jedes Medizinprodukt einer bestimmten Klasse zugeordnet werden muss. Von dieser Zuordnung hängt das weitere
Nicht Invasiv. Regel 1-4. D:\flash_work\Klassifizierung von MP\DOC\flow_chart_1.odg Version 2.5 Nicht Invasiv Andreas Hilburg
 Nicht nvasiv Regel 1-4 Start 0.0.0 Produkt invasiv? Dringt das Produkt, durch die Körperoberfläche oder über eine Körperöffnung ganz oder teilweise in den Körper ein? 1.1.0 2.0.0 Regel 2 Produkt für die
Nicht nvasiv Regel 1-4 Start 0.0.0 Produkt invasiv? Dringt das Produkt, durch die Körperoberfläche oder über eine Körperöffnung ganz oder teilweise in den Körper ein? 1.1.0 2.0.0 Regel 2 Produkt für die
Abschnitt 1 Anwendungsbereich und Allgemeine Anforderungen an die Konformitätsbewertung 1 Anwendungsbereich
 13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") 05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
Keine CE-Kennzeichnung ohne klinische Bewertung
 Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Klassifizierungsregeln bei Medizinprodukten
 bei Medizinprodukten Unter Verwendung von Material von Dr. Johann Rader TÜV SÜD Product Service GmbH, München 1/56 Regel 1 (nicht-invasive Produkte) Regel 1 (nicht-invasive Produkte) Alle nicht invasiven
bei Medizinprodukten Unter Verwendung von Material von Dr. Johann Rader TÜV SÜD Product Service GmbH, München 1/56 Regel 1 (nicht-invasive Produkte) Regel 1 (nicht-invasive Produkte) Alle nicht invasiven
Medizintechnik: Regulatorische Rahmenbedingungen I
 Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Dr. Cornelia Henschke FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Dr. Cornelia Henschke FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
Verordnung über Medizinprodukte (Medizinprodukte- Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I. S. 3854) Auf Grund des 37 Abs. 1, 8 und 11 des Medizinproduktegesetzes vom 2. August 1994 (BGBl. I. S.
BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH. Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II
 1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
Medizintechnik: Regulatorische Rahmenbedingungen I
 Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Prof. Dr. med Reinhard Busse FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Prof. Dr. med Reinhard Busse FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
MEDIZINPRODUKTEABGABENVERORDNUNG
 MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
MEDIZINPRODUKTEABGABENVERORDNUNG Wer ist abgabepflichtig? Jede natürliche und juristische Person, die Medizinprodukte an Letztverbraucher entgeltlich abgibt. Unter Abgeben ist in diesem Fall die entgeltliche
Medizintechnik: Regulatorische Rahmenbedingungen I
 Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Prof. Dr. med Reinhard Busse FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
Management im Gesundheitswesen III Industrie Medizintechnik: Regulatorische Rahmenbedingungen I Prof. Dr. med Reinhard Busse FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating
Software. Martin Zauner. martin.zauner@fh-linz.at +43 (0)732 804 52100. FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz
732 804 52100. FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz") Martin Zauner martin.zauner@fh-linz.at +43 (0)732 804 52100 FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz 1 Gesundheitswesen Zusammenwachsen zweier Welten bringt neue Herausforderungen
Martin Zauner martin.zauner@fh-linz.at +43 (0)732 804 52100 FH Oberösterreich - Studiengang Medizintechnik Garnisonstrasse 21 A 4020 Linz 1 Gesundheitswesen Zusammenwachsen zweier Welten bringt neue Herausforderungen
Verordnung zur Änderung medizinprodukterechtlicher Vorschriften vom 16. Februar 2007
 26.02.2007 Verordnung zur Änderung medizinprodukterechtlicher Vorschriften vom 16. Februar 2007 Auf Grund des 37 Abs. 1, 9, 10 und 11 Satz 1 des Medizinproduktegesetzes in der Fassung der Bekanntmachung
26.02.2007 Verordnung zur Änderung medizinprodukterechtlicher Vorschriften vom 16. Februar 2007 Auf Grund des 37 Abs. 1, 9, 10 und 11 Satz 1 des Medizinproduktegesetzes in der Fassung der Bekanntmachung
Medizinprodukte-Sicherheitsplanverordnung
 Hygieneforum Sande 19.06.2014 Meldung von Vorkommnissen nach der Medizinproduke-Sicherheitsplanverordnung www.klinikum-bremen-ldw.de Klinikum Links der Weser Senator-Weßling-Str. 1 28277 Bremen Meldung
Hygieneforum Sande 19.06.2014 Meldung von Vorkommnissen nach der Medizinproduke-Sicherheitsplanverordnung www.klinikum-bremen-ldw.de Klinikum Links der Weser Senator-Weßling-Str. 1 28277 Bremen Meldung
MPG und VO zum 94. N. Buchrieser
 MPG und VO zum 94 N. Buchrieser Gesetze und Regelwerke EU-Direktive 93/42/EWG Medizinproduktegesetz Verordnung zum 94 MPG (ÖGSV-LL 11) RDG-Normen (ÖNORM EN ISO 15883 Teil 1-7) Sterilisatornormen (EN 285,
MPG und VO zum 94 N. Buchrieser Gesetze und Regelwerke EU-Direktive 93/42/EWG Medizinproduktegesetz Verordnung zum 94 MPG (ÖGSV-LL 11) RDG-Normen (ÖNORM EN ISO 15883 Teil 1-7) Sterilisatornormen (EN 285,
Open Source Software als Medizinprodukt
 Open Source Software als Medizinprodukt 1 Begrüßung Innovativer PACS-Hersteller seit 1996 seit 2005 im OsiriX Projekt dabei Marktführer im DICOM Paperprinting 2 Themen Open Source Software (OSS) OsiriX
Open Source Software als Medizinprodukt 1 Begrüßung Innovativer PACS-Hersteller seit 1996 seit 2005 im OsiriX Projekt dabei Marktführer im DICOM Paperprinting 2 Themen Open Source Software (OSS) OsiriX
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten
 Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Regulatorische Anforderungen an die Entwicklung von Medizinprodukten Alexander Fink, Metecon GmbH Institut für Medizintechnik Reutlingen University Alteburgstraße 150 D-72762 Reutlingen Reutlingen, 04.03.2015
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) MPV Ausfertigungsdatum: 20.12.2001 Vollzitat: "Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I S. 3854), die zuletzt durch Artikel
Medizinprodukte mit pflanzlichen Inhaltsstoffen - eine Alternative zur Arzneimittelzulassung
 Medizinprodukte mit pflanzlichen Inhaltsstoffen - eine Alternative zur Arzneimittelzulassung Dr. Nicole Armbrüster 20. Bernburger Winterseminar 23./24. Februar 2010 analyze & realize ag Waldseeweg 13467
Medizinprodukte mit pflanzlichen Inhaltsstoffen - eine Alternative zur Arzneimittelzulassung Dr. Nicole Armbrüster 20. Bernburger Winterseminar 23./24. Februar 2010 analyze & realize ag Waldseeweg 13467
Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar?
 Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar? 04.06.2013 Medical Apps 2013 Kathrin Schürmann, Rechtsanwältin 1 2013 ISiCO Datenschutz GmbH All rights reserved 2 1 Chancen und Risiken
Stellen Gesundheits- und Medizin Apps ein Sicherheitsrisiko dar? 04.06.2013 Medical Apps 2013 Kathrin Schürmann, Rechtsanwältin 1 2013 ISiCO Datenschutz GmbH All rights reserved 2 1 Chancen und Risiken
1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene Anwendung über einen Zeitraum von weniger als 60 Minuten bestimmt.
 Klassifizierung gemäß 93/42/EWG ANHANG IX KLASSIFIZIERUNGSKRITERIEN I. DEFINITONEN 1. Definitionen zu den Klassifizierungsregeln 1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene
Klassifizierung gemäß 93/42/EWG ANHANG IX KLASSIFIZIERUNGSKRITERIEN I. DEFINITONEN 1. Definitionen zu den Klassifizierungsregeln 1.1. Dauer Vorübergehend: Unter normalen Bedingungen für eine ununterbrochene
Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren
 Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit 1 Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren Politische Ebene Bundesbehörden Ministerien, insbesondere
Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit 1 Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren Politische Ebene Bundesbehörden Ministerien, insbesondere
Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext
 Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext Reinhard Busse, Prof. Dr. med. MPH FFPH Fachgebiet Management im Gesundheitswesen, Technische Universität Berlin (WHO
Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext Reinhard Busse, Prof. Dr. med. MPH FFPH Fachgebiet Management im Gesundheitswesen, Technische Universität Berlin (WHO
Alles tot geregelt im Medizinprodukterecht? Was möchte der Gesetzgeber erreichen? Was riskiere ich bei Nichtbeachtung?
 Alles tot geregelt im Medizinprodukterecht? Was möchte der Gesetzgeber erreichen? Was riskiere ich bei Nichtbeachtung? Ein Vortrag von Dipl.-Ing. Thomas J. Pleiss, öffentlich bestellter und vereidigter
Alles tot geregelt im Medizinprodukterecht? Was möchte der Gesetzgeber erreichen? Was riskiere ich bei Nichtbeachtung? Ein Vortrag von Dipl.-Ing. Thomas J. Pleiss, öffentlich bestellter und vereidigter
26. Sitzung des Gesundheitsforschungsrates (GFR) am 12. Dezember 2008. Entschließung. Klinische Studien in der Medizintechnik
 am 12. Dezember 2008. Entschließung. Klinische Studien in der Medizintechnik") 26. Sitzung des Gesundheitsforschungsrates (GFR) am 12. Dezember 2008 Entschließung Klinische Studien in der Medizintechnik Auf Anregung seines Medizintechnischen Ausschusses hat der Gesundheitsforschungsrat
26. Sitzung des Gesundheitsforschungsrates (GFR) am 12. Dezember 2008 Entschließung Klinische Studien in der Medizintechnik Auf Anregung seines Medizintechnischen Ausschusses hat der Gesundheitsforschungsrat
für die Aufbereitung von Medizinprodukten
 Die Bedeutung der Biokompatibilität für die Aufbereitung von Medizinprodukten Dr.-Ing. Ute Müller, Geschäftsführer BMP Labor für medizinische Materialprüfung GmbH, Aachen Inhalt Einordnung der Biokompatibilität
Die Bedeutung der Biokompatibilität für die Aufbereitung von Medizinprodukten Dr.-Ing. Ute Müller, Geschäftsführer BMP Labor für medizinische Materialprüfung GmbH, Aachen Inhalt Einordnung der Biokompatibilität
8.5. Medizinproduktegesetz
 8.5. Medizinproduktegesetz 1 Zweck des Gesetzes http://bundesrecht.juris.de/bundesrecht/mpg/ 3 Begriffsbestimmungen Zweck dieses Gesetzes ist es, den Verkehr mit Medizinprodukten zu regeln und dadurch
8.5. Medizinproduktegesetz 1 Zweck des Gesetzes http://bundesrecht.juris.de/bundesrecht/mpg/ 3 Begriffsbestimmungen Zweck dieses Gesetzes ist es, den Verkehr mit Medizinprodukten zu regeln und dadurch
Biologische Sicherheitsprüfungen nach DIN EN ISO 10993
 Biologische Sicherheitsprüfungen nach DIN EN ISO 10993 Referentin: Kontakt: Mirjam Ruess Mirjam.Ruess@dqs.de Seite 1/ 10-2009 Inhalt 1. Klassifizierung von Medizinprodukten 2. Regulatorische Vorgaben 3.
Biologische Sicherheitsprüfungen nach DIN EN ISO 10993 Referentin: Kontakt: Mirjam Ruess Mirjam.Ruess@dqs.de Seite 1/ 10-2009 Inhalt 1. Klassifizierung von Medizinprodukten 2. Regulatorische Vorgaben 3.
EUROPÄISCHE KOMMISSION
 24.1.2013 Amtsblatt der Europäischen Union C 22/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
24.1.2013 Amtsblatt der Europäischen Union C 22/1 IV (Informationen) INFORMATIONEN DER ORGANE, EINRICHTUNGEN UND SONSTIGEN STELLEN DER EUROPÄISCHEN UNION EUROPÄISCHE KOMMISSION Mitteilung der Kommission
InVo. Information zu Verordnungen in der GKV. Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen. Stand: Februar 2010
 Nr. 1 2010 InVo Information zu Verordnungen in der GKV Stand: Februar 2010 Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen Bisher konnten Sie als Arzt Arzneimittel (z. B.
Nr. 1 2010 InVo Information zu Verordnungen in der GKV Stand: Februar 2010 Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen Bisher konnten Sie als Arzt Arzneimittel (z. B.
MDD Recast Report. Stand 30. Januar 2013. Rafael J. de la Roza
 MDD Recast Report Die geplanten Änderungen des europäischen Rechtsrahmens für Medizinprodukte und die Änderungen für Hersteller, Bevollmächtigte, Importeure und den Handel Stand 30. Januar 2013 Rafael
MDD Recast Report Die geplanten Änderungen des europäischen Rechtsrahmens für Medizinprodukte und die Änderungen für Hersteller, Bevollmächtigte, Importeure und den Handel Stand 30. Januar 2013 Rafael
Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten
 Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten Alexander Fink Hochschule Mannheim SS 2013 A. Fink Seite 1 Steigen wir ein Wo und wie kommt der Medizintechnik-Ingenieur
Anforderungen der Medizinprodukte- Richtlinie an die Entwicklung von Medizinprodukten Alexander Fink Hochschule Mannheim SS 2013 A. Fink Seite 1 Steigen wir ein Wo und wie kommt der Medizintechnik-Ingenieur
Medizinprodukte - was Hersteller und Händler wissen sollten
 Medizinprodukte - was Hersteller und Händler wissen sollten I. Medizinprodukte ein Überblick 1. Anwendungsbereich des Gesetzes über Medizinprodukte Schon der Begriff der Medizinprodukte ist nicht leicht
Medizinprodukte - was Hersteller und Händler wissen sollten I. Medizinprodukte ein Überblick 1. Anwendungsbereich des Gesetzes über Medizinprodukte Schon der Begriff der Medizinprodukte ist nicht leicht
Unser Leitbild. Medizinprodukte. Unverzichtbar für das Leben. Interessensvertretung der Medizinprodukte-Unternehmen
 Unser Leitbild. Medizinprodukte. Unverzichtbar für das Leben. Interessensvertretung der Medizinprodukte-Unternehmen Austromed UNSER Leitbild 1. AUSTROMED UNSERE MISSION 2. AUSTROMED MEDIZINPRODUKTE SIND
Unser Leitbild. Medizinprodukte. Unverzichtbar für das Leben. Interessensvertretung der Medizinprodukte-Unternehmen Austromed UNSER Leitbild 1. AUSTROMED UNSERE MISSION 2. AUSTROMED MEDIZINPRODUKTE SIND
Erläuterungen zur Untervergabe von Instandhaltungsfunktionen
 Zentrale Erläuterungen zur Untervergabe von Instandhaltungsfunktionen Gemäß Artikel 4 der Verordnung (EU) 445/2011 umfasst das Instandhaltungssystem der ECM die a) Managementfunktion b) Instandhaltungsentwicklungsfunktion
Zentrale Erläuterungen zur Untervergabe von Instandhaltungsfunktionen Gemäß Artikel 4 der Verordnung (EU) 445/2011 umfasst das Instandhaltungssystem der ECM die a) Managementfunktion b) Instandhaltungsentwicklungsfunktion
STUDY NURSE AKADEMIE der AGAH e.v. Das neue Medizinproduktegesetz: Was hat es gebracht? 31.05.2013 Dr. Ralf Schweitzer
 STUDY NURSE AKADEMIE der AGAH e.v. Das neue Medizinproduktegesetz: Was hat es gebracht? 31.05.2013 Dr. Ralf Schweitzer Darum wird es in diesem Teil gehen Medizinprodukte Klinische Prüfungen von Medizinprodukten
STUDY NURSE AKADEMIE der AGAH e.v. Das neue Medizinproduktegesetz: Was hat es gebracht? 31.05.2013 Dr. Ralf Schweitzer Darum wird es in diesem Teil gehen Medizinprodukte Klinische Prüfungen von Medizinprodukten
PLM: Privat Label Manufacturer (Kunde der OEM-PLM-Beziehung) OEM: Original Equipment Manufacturer (Lieferant der OEM-PLM-Beziehung)
 OEM: Original Equipment Manufacturer (Lieferant der OEM-PLM-Beziehung)") 1 Anwendsbereich Prüf der Produktdokumentation im Falle von Privat Label Herstellern gemäß OEM Verfahren Zu verwenden bei neuen Anträgen zur Zertifizier und bei Stichprobenprüfen im Rahmen der regelmäßigen
1 Anwendsbereich Prüf der Produktdokumentation im Falle von Privat Label Herstellern gemäß OEM Verfahren Zu verwenden bei neuen Anträgen zur Zertifizier und bei Stichprobenprüfen im Rahmen der regelmäßigen
Entwurf eines Gesetzes zur Änderung medizinprodukterechtlicher Vorschriften
 Deutscher Bundestag Drucksache 16/12258 16. Wahlperiode 16. 03. 2009 Gesetzentwurf der Bundesregierung Entwurf eines Gesetzes zur Änderung medizinprodukterechtlicher Vorschriften A. Problem und Ziel DiesesGesetzdientvornehmlichderUmsetzungderRichtlinie2007/47/EGdes
Deutscher Bundestag Drucksache 16/12258 16. Wahlperiode 16. 03. 2009 Gesetzentwurf der Bundesregierung Entwurf eines Gesetzes zur Änderung medizinprodukterechtlicher Vorschriften A. Problem und Ziel DiesesGesetzdientvornehmlichderUmsetzungderRichtlinie2007/47/EGdes
Software als Medizinprodukt
 Software als Medizinprodukt DI Dr. Gerhard Wrodnigg, MSc. TÜV AUSTRIA SERVICES Software als Medizinprodukt Wann ist Software ein Medizinprodukt? Änderung der RL 93/42/EWG durch 2007/47/EG Qualification
Software als Medizinprodukt DI Dr. Gerhard Wrodnigg, MSc. TÜV AUSTRIA SERVICES Software als Medizinprodukt Wann ist Software ein Medizinprodukt? Änderung der RL 93/42/EWG durch 2007/47/EG Qualification
Know-How für die Medizintechnik
 CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
CMI-WORKSHOP Know-How für die Medizintechnik Medizinprodukte Klasse I Vademecum für den Marktzugang Grundlegende Anforderungen Harmonisierte Normen Technische Dokumentation Regulatory Compliance Solutions
Sicherheit von Patientinnen und Patienten bei Medizinprodukten
 Deutscher Bundestag Drucksache 17/9009 17. Wahlperiode 19. 03. 2012 Antwort der Bundesregierung auf die Kleine Anfrage der Abgeordneten Dr. Martina Bunge, Kathrin Vogler, Diana Golze, weiterer Abgeordneter
Deutscher Bundestag Drucksache 17/9009 17. Wahlperiode 19. 03. 2012 Antwort der Bundesregierung auf die Kleine Anfrage der Abgeordneten Dr. Martina Bunge, Kathrin Vogler, Diana Golze, weiterer Abgeordneter
Neuerungen im Medizinprodukterecht Konsequenzen für Hersteller
 Neuerungen im Medizinprodukterecht Konsequenzen für Hersteller Dr. Michael Rinck ZPT St.Ingbert, 04. Juni 2009 Übersicht Änderung der gesetzlichen Regelungen in Europa Medizinprodukte Angrenzende Bereiche
Neuerungen im Medizinprodukterecht Konsequenzen für Hersteller Dr. Michael Rinck ZPT St.Ingbert, 04. Juni 2009 Übersicht Änderung der gesetzlichen Regelungen in Europa Medizinprodukte Angrenzende Bereiche
Zertifizierung des QM-Systems bei der Aufbereitung von Kritisch-C Produkten
 Christian Witte Zertifizierung des QM-Systems bei der Aufbereitung von Kritisch-C Produkten Zur aktuellen Differenz zwischen MPBetreibV und KRINKO/BfArM-Empfehlung Verordnung über die Änderung der MPBetreibV
Christian Witte Zertifizierung des QM-Systems bei der Aufbereitung von Kritisch-C Produkten Zur aktuellen Differenz zwischen MPBetreibV und KRINKO/BfArM-Empfehlung Verordnung über die Änderung der MPBetreibV
Was ist für den Anwender interessant und wie kommen Produkte auf den Markt?
 Was ist für den Anwender interessant und wie kommen Produkte auf den Markt? 3. Workshop Neue Horizonte für metallische Biomaterialien http://medalt.h-e-s.de/image/image_gallery?img_id=30298747 http://www.ifam-dd.fraunhofer.de/de/zellulare_metallische_werkstoffe/offenzellige_metallschaeume/technologie/biomaterialien/werkstoffe/_jcr_content/stage/image.img.jpg/1317812850007.jpg
Was ist für den Anwender interessant und wie kommen Produkte auf den Markt? 3. Workshop Neue Horizonte für metallische Biomaterialien http://medalt.h-e-s.de/image/image_gallery?img_id=30298747 http://www.ifam-dd.fraunhofer.de/de/zellulare_metallische_werkstoffe/offenzellige_metallschaeume/technologie/biomaterialien/werkstoffe/_jcr_content/stage/image.img.jpg/1317812850007.jpg
Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt?
 DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
Anlage zur Akkreditierungsurkunde D-ZE-16029-04-00 nach DIN EN ISO/IEC 17065:2013
 Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZE-16029-04-00 nach DIN EN ISO/IEC 17065:2013 Gültigkeitsdauer: 27.04.2016 bis 14.12.2019 Ausstellungsdatum: 27.04.2016 Urkundeninhaber:
Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZE-16029-04-00 nach DIN EN ISO/IEC 17065:2013 Gültigkeitsdauer: 27.04.2016 bis 14.12.2019 Ausstellungsdatum: 27.04.2016 Urkundeninhaber:
MEDIZINPRODUKTE. Chancen und Herausforderungen aus Sicht der Industrie
 MEDIZINPRODUKTE Chancen und Herausforderungen aus Sicht der Industrie Bonn, 16. September 2015, BfArM, Bundesinstitut für Arzneimittel und Medizinprodukte Joachim M. Schmitt Geschäftsführer/Vorstandsmitglied
MEDIZINPRODUKTE Chancen und Herausforderungen aus Sicht der Industrie Bonn, 16. September 2015, BfArM, Bundesinstitut für Arzneimittel und Medizinprodukte Joachim M. Schmitt Geschäftsführer/Vorstandsmitglied
Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen
 Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen Risiken erkennen, minimieren, finanzieren heute und morgen SAQ Fachgruppe Medizinprodukte Hersteller, 22.3.2007, Zofingen Monika Gattiker
Die Risiken von morgen: Anpassungen der EG-Richtlinien und ihre Folgen Risiken erkennen, minimieren, finanzieren heute und morgen SAQ Fachgruppe Medizinprodukte Hersteller, 22.3.2007, Zofingen Monika Gattiker
Anhang V EG-Konformitätserklärung (Qualitätssicherung Produktion)
") Dieses Werk, einschließlich aller seiner Teile, ist urheberrechtlich geschützt. Jede Verwertung außerhalb der engen Grenzen des Urheberrechtsgesetzes ist ohne Zustimmung des Verlages unzulässig und strafbar.
Dieses Werk, einschließlich aller seiner Teile, ist urheberrechtlich geschützt. Jede Verwertung außerhalb der engen Grenzen des Urheberrechtsgesetzes ist ohne Zustimmung des Verlages unzulässig und strafbar.
Nominative und gesetzliche Grundlagen zur Aufbereitung von Endoskopen
 Nominative und gesetzliche Grundlagen zur Aufbereitung von Endoskopen Dr. med. univ. Sebastian Werner 1,2 1 HygCen Austria GmbH, Bischofshofen 2 Abteilung für Hygiene, Sozial und Umweltmedizin, Ruhruniversität
Nominative und gesetzliche Grundlagen zur Aufbereitung von Endoskopen Dr. med. univ. Sebastian Werner 1,2 1 HygCen Austria GmbH, Bischofshofen 2 Abteilung für Hygiene, Sozial und Umweltmedizin, Ruhruniversität
Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext
 Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext Reinhard Busse, Prof. Dr. med. MPH FFPH Fachgebiet Management im Gesundheitswesen, Technische Universität Berlin (WHO
Finanzierung von Medizintechnikprodukten in Deutschland im internationalen Kontext Reinhard Busse, Prof. Dr. med. MPH FFPH Fachgebiet Management im Gesundheitswesen, Technische Universität Berlin (WHO
Zur Wahrung der Widerrufsfrist reicht es aus, dass Sie die Mitteilung über die Ausübung des Widerrufsrechts vor Ablauf der Widerrufsfrist absenden.
 Widerrufsbelehrung der Firma Widerrufsbelehrung - Verträge für die Lieferung von Waren Ist der Kunde Unternehmer ( 14 BGB), so hat er kein Widerrufs- und Rückgaberecht gem. 312g BGB i. V. m. 355 BGB. Das
Widerrufsbelehrung der Firma Widerrufsbelehrung - Verträge für die Lieferung von Waren Ist der Kunde Unternehmer ( 14 BGB), so hat er kein Widerrufs- und Rückgaberecht gem. 312g BGB i. V. m. 355 BGB. Das
Kundenfragebogen zur Zertifizierung von Managementsystemen
 Bitte beantworten Sie die folgenden Fragen und senden Sie uns diesen Fragebogen per Fax (0316/873-107395) oder Mail (pmg@tugraz.at) retour. 1. Allgemeine Informationen Allgemeine Angaben zum Unternehmen:
Bitte beantworten Sie die folgenden Fragen und senden Sie uns diesen Fragebogen per Fax (0316/873-107395) oder Mail (pmg@tugraz.at) retour. 1. Allgemeine Informationen Allgemeine Angaben zum Unternehmen:
Ergänzende Schutzzertifikate für Medizinische Geräte/Medizinprodukte?
 Ergänzende Schutzzertifikate für Medizinische Geräte/Medizinprodukte? Dr. Matthias Kindler 1 Erzeugnis Zulassung CE-Zertifizierung gleichgestellt zu Arzneimittelzulassung gemäß Richtlinie 2001/83/EG SPC
Ergänzende Schutzzertifikate für Medizinische Geräte/Medizinprodukte? Dr. Matthias Kindler 1 Erzeugnis Zulassung CE-Zertifizierung gleichgestellt zu Arzneimittelzulassung gemäß Richtlinie 2001/83/EG SPC
Rechtliche Aspekte der Aufbereitung von Medizinprodukten
 IG NOOPS 08.06.2012 Rechtliche Aspekte der Aufbereitung von Medizinprodukten Andrea Schütz Frikart Inspektor Marktkontrolle Medizinprodukte andrea.schuetzfrikart@swissmedic.ch Swissmedic Schweizerisches
IG NOOPS 08.06.2012 Rechtliche Aspekte der Aufbereitung von Medizinprodukten Andrea Schütz Frikart Inspektor Marktkontrolle Medizinprodukte andrea.schuetzfrikart@swissmedic.ch Swissmedic Schweizerisches
(Veröffentlichung der Titel und der Bezugsnummern der harmonisierten Normen im Sinne der Harmonisierungsrechtsvorschriften
 C 14/36 DE Amtsblatt der Europäischen Union 16.1.2015 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschriften der
C 14/36 DE Amtsblatt der Europäischen Union 16.1.2015 Mitteilung der Kommission im Rahmen der Durchführung der Richtlinie 90/385/EWG des Rates vom 20. Juni 1990 zur Angleichung der Rechtsvorschriften der
Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de
 ISO/IEC 62304 Medizingeräte-Software Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de DQS Medizin nprodukte GmbH Übersicht Basics Wann ist ein MP Software? Markteinführung vor der 62304 alles
ISO/IEC 62304 Medizingeräte-Software Referent: Mathias Notheis Kontakt: Mathias.Notheis@dqs.de DQS Medizin nprodukte GmbH Übersicht Basics Wann ist ein MP Software? Markteinführung vor der 62304 alles
Das BASG / AGES PharmMed
 CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
Merkblatt. Häufige Fragen hinsichtlich der Anforderungen für Hersteller bzw. Inverkehrbringer von Lebensmittelbedarfsgegenständen aus Keramik
 Merkblatt Häufige Fragen hinsichtlich der Anforderungen für Hersteller bzw. Inverkehrbringer von Lebensmittelbedarfsgegenständen aus Keramik Was sind Lebensmittelbedarfsgegenstände? Lebensmittelbedarfsgegenstände
Merkblatt Häufige Fragen hinsichtlich der Anforderungen für Hersteller bzw. Inverkehrbringer von Lebensmittelbedarfsgegenständen aus Keramik Was sind Lebensmittelbedarfsgegenstände? Lebensmittelbedarfsgegenstände
RKI-Empfehlung/Risikobewertung 11.1
 Anhang 1 Unkritische Medizinprodukte Medizinprodukte, die lediglich mit intakter Haut in Unkritische Medizinprodukte z.b. Instrumente für Maßnahmen ohne Schleimhautkontakt (z.b. extraorale Teile des Gesichtsbogens,
Anhang 1 Unkritische Medizinprodukte Medizinprodukte, die lediglich mit intakter Haut in Unkritische Medizinprodukte z.b. Instrumente für Maßnahmen ohne Schleimhautkontakt (z.b. extraorale Teile des Gesichtsbogens,
Risikomanagement bei Medizinprodukten
 Risikomanagement bei Medizinprodukten 10. Jahrestagung der AAL 24./25. September 2010 Stuttgart 2010 mdc medical device certification GmbH Risikomanagement 1 Regulatorische Grundlagen Richtlinie 93/42/EWG
Risikomanagement bei Medizinprodukten 10. Jahrestagung der AAL 24./25. September 2010 Stuttgart 2010 mdc medical device certification GmbH Risikomanagement 1 Regulatorische Grundlagen Richtlinie 93/42/EWG
Softwarevalidierung aus Anwendersicht. DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009
 Softwarevalidierung aus Anwendersicht DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009 Softwarevalidierung aus Anwendersicht Geräte mit automatischen Prozessabläufen zur Aufbereitung von Medizinprodukten
Softwarevalidierung aus Anwendersicht DGSV Kongress / Dr. B. Gallert / Fulda / 16.10.2009 Softwarevalidierung aus Anwendersicht Geräte mit automatischen Prozessabläufen zur Aufbereitung von Medizinprodukten
REACH im Überblick - Stand der Umsetzung Dr. Raimund Weiß Hamburg 19. März 2015
 REACH im Überblick - Stand der Umsetzung Dr. Raimund Weiß Hamburg 19. März 2015 Inhalt Zahlen SVHC Roadmap national Erzeugnisse Zulassung Bedeutung von REACH REACH ist eine europäische Verordnung, die
REACH im Überblick - Stand der Umsetzung Dr. Raimund Weiß Hamburg 19. März 2015 Inhalt Zahlen SVHC Roadmap national Erzeugnisse Zulassung Bedeutung von REACH REACH ist eine europäische Verordnung, die
Rückverfolgbarkeit von Lebensmitteln Erfahrungen aus den Ländern
 Rückverfolgbarkeit von Lebensmitteln Erfahrungen aus den Ländern Untersuchung und Erfassung lebensmittelbedingter Ausbrüche Informationsveranstaltung des Bundesinstituts für Risikobewertung am 25. Januar
Rückverfolgbarkeit von Lebensmitteln Erfahrungen aus den Ländern Untersuchung und Erfassung lebensmittelbedingter Ausbrüche Informationsveranstaltung des Bundesinstituts für Risikobewertung am 25. Januar
Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege
 Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege. Dipl. Ing. Norbert Kamps Referent für Hilfsmittelversorgung Medizinischer Dienst des Spitzenverbandes Bund der Krankenkassen e.v. Fachgebiet
Medizinproduktegesetz Auswirkungen und Bedeutung für die Pflege. Dipl. Ing. Norbert Kamps Referent für Hilfsmittelversorgung Medizinischer Dienst des Spitzenverbandes Bund der Krankenkassen e.v. Fachgebiet
DÜNGEMITTELRECHTLICHE ASPEKTE BÖDEN. Hans-Walter Schneichel Struktur- und Genehmigungsdirektion Nord, Koblenz
 DÜNGEMITTELRECHTLICHE ASPEKTE DESEINSATZESVON EINSATZES BIOKOHLE IN BÖDEN Berlin, den 05.10.2011 Hans-Walter Schneichel Struktur- und Genehmigungsdirektion Nord, Koblenz Das Aufbringen und Einbringen von
DÜNGEMITTELRECHTLICHE ASPEKTE DESEINSATZESVON EINSATZES BIOKOHLE IN BÖDEN Berlin, den 05.10.2011 Hans-Walter Schneichel Struktur- und Genehmigungsdirektion Nord, Koblenz Das Aufbringen und Einbringen von
Diese Broschüre fasst die wichtigsten Informationen zusammen, damit Sie einen Entscheid treffen können.
 Aufklärung über die Weiterverwendung/Nutzung von biologischem Material und/oder gesundheitsbezogen Daten für die biomedizinische Forschung. (Version V-2.0 vom 16.07.2014, Biobanken) Sehr geehrte Patientin,
Aufklärung über die Weiterverwendung/Nutzung von biologischem Material und/oder gesundheitsbezogen Daten für die biomedizinische Forschung. (Version V-2.0 vom 16.07.2014, Biobanken) Sehr geehrte Patientin,
Anlage zur Akkreditierungsurkunde D-ZM-16002-06-00 nach DIN EN ISO/IEC 17021:2011
 Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-16002-06-00 nach DIN EN ISO/IEC 17021:2011 Gültigkeitsdauer: 15.12.2014 bis 14.12.2019 Ausstellungsdatum: 15.12.2014 Urkundeninhaber:
Deutsche Akkreditierungsstelle GmbH Anlage zur Akkreditierungsurkunde D-ZM-16002-06-00 nach DIN EN ISO/IEC 17021:2011 Gültigkeitsdauer: 15.12.2014 bis 14.12.2019 Ausstellungsdatum: 15.12.2014 Urkundeninhaber:
6 Informationsermittlung und Gefährdungsbeurteilung
 Verordnung zum Schutz vor Gefahrstoffen TK Lexikon Arbeitsrecht 6 Informationsermittlung und Gefährdungsbeurteilung HI2516431 (1) 1 Im Rahmen einer Gefährdungsbeurteilung als Bestandteil der Beurteilung
Verordnung zum Schutz vor Gefahrstoffen TK Lexikon Arbeitsrecht 6 Informationsermittlung und Gefährdungsbeurteilung HI2516431 (1) 1 Im Rahmen einer Gefährdungsbeurteilung als Bestandteil der Beurteilung
Regulative Aspekte der Aufbereitung chirurgischer Implantate
 Regulative Aspekte der Aufbereitung chirurgischer Implantate 35.Veranstaltung des Arbeitskreis Infektionsprophylaxe 16. Oktober 2012 in Potsdam 17.Oktober 2012 in Leipzig 1 Person Anja Fechner Dipl.-Ing.
Regulative Aspekte der Aufbereitung chirurgischer Implantate 35.Veranstaltung des Arbeitskreis Infektionsprophylaxe 16. Oktober 2012 in Potsdam 17.Oktober 2012 in Leipzig 1 Person Anja Fechner Dipl.-Ing.
Exkurs: Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen
 Exkurs: Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen 139a SGB V Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (1) Der Gemeinsame Bundesausschuss nach 91 gründet
Exkurs: Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen 139a SGB V Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (1) Der Gemeinsame Bundesausschuss nach 91 gründet
312a Allgemeine Pflichten und Grundsätze bei Verbraucherverträgen; Grenzen der Vereinbarung von Entgelten
 312a Allgemeine Pflichten und Grundsätze bei Verbraucherverträgen; Grenzen der Vereinbarung von Entgelten (1) Ruft der Unternehmer oder eine Person, die in seinem Namen oder Auftrag handelt, den Verbraucher
312a Allgemeine Pflichten und Grundsätze bei Verbraucherverträgen; Grenzen der Vereinbarung von Entgelten (1) Ruft der Unternehmer oder eine Person, die in seinem Namen oder Auftrag handelt, den Verbraucher
Europäische Technische Bewertung. ETA-14/0212 vom 27. Juni 2014. Allgemeiner Teil
 Europäische Technische Bewertung ETA-14/0212 vom 27. Juni 2014 Allgemeiner Teil Technische Bewertungsstelle, die die Europäische Technische Bewertung ausstellt Handelsname des Bauprodukts Produktfamilie,
Europäische Technische Bewertung ETA-14/0212 vom 27. Juni 2014 Allgemeiner Teil Technische Bewertungsstelle, die die Europäische Technische Bewertung ausstellt Handelsname des Bauprodukts Produktfamilie,
Warum? Orthopäden; 5336. Dermatologen; 3342. Urologen; 2674. Chirurgen; 3976. Neurochirurgen; 491. Gynäkologen; 9875
 Können Medizinprodukte ohne CE-Kennzeichnung betrieben werden? Warum? Urologen; 2674 Neurochirurgen; 491 MKG; 1027 Allgemeinärzte/ praktische Ärzte; 40.246 Orthopäden; 5336 Chirurgen; 3976 Anästhesisten;
Können Medizinprodukte ohne CE-Kennzeichnung betrieben werden? Warum? Urologen; 2674 Neurochirurgen; 491 MKG; 1027 Allgemeinärzte/ praktische Ärzte; 40.246 Orthopäden; 5336 Chirurgen; 3976 Anästhesisten;
Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium
 QUALITY-APPS Applikationen für das Qualitätsmanagement Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium Autor: Prof. Dr. Jürgen P. Bläsing Die Maschinenrichtlinie 2006/42/EG ist
QUALITY-APPS Applikationen für das Qualitätsmanagement Maschinenrichtlinie 2006/42/EG 150 Fragen und Antworten zum Selbststudium Autor: Prof. Dr. Jürgen P. Bläsing Die Maschinenrichtlinie 2006/42/EG ist
CE-Kennzeichnung bei Medizinprodukten- Grundlagen und Rahmenbedingungen
 CE-Kennzeichnung bei Medizinprodukten- Grundlagen und Rahmenbedingungen Dr. Heike Wachenhausen, Rechtsanwältin IHK zu Lübeck, 19. Juni 2015 Übersicht I. Verkehrsfähigkeit von Medizinprodukten II. III.
CE-Kennzeichnung bei Medizinprodukten- Grundlagen und Rahmenbedingungen Dr. Heike Wachenhausen, Rechtsanwältin IHK zu Lübeck, 19. Juni 2015 Übersicht I. Verkehrsfähigkeit von Medizinprodukten II. III.
1.1 Inhaltsverzeichnis
 Verzeichnisse Inhaltsverzeichnis 1.1 1.1 Inhaltsverzeichnis 1 Verzeichnisse 1.1 Inhaltsverzeichnis 1.2 Herausgeber- und Autorenverzeichnis 1.3 Stichwortverzeichnis 2 Aktuelles 2.1 Änderungen des europäischen
Verzeichnisse Inhaltsverzeichnis 1.1 1.1 Inhaltsverzeichnis 1 Verzeichnisse 1.1 Inhaltsverzeichnis 1.2 Herausgeber- und Autorenverzeichnis 1.3 Stichwortverzeichnis 2 Aktuelles 2.1 Änderungen des europäischen
Dr. Thomas Mall Donnerstag, 29. Januar 2015. Update Neue IVD-Verordnung
 Dr. Thomas Mall Update Neue IVD-Verordnung Wo stehen wir? Vorschlag der EU-Kommission für eine Verordnung über In-vitro- Diagnostika am 26.09.2012 veröffentlicht Parallele Veröffentlichung des Vorschlages
Dr. Thomas Mall Update Neue IVD-Verordnung Wo stehen wir? Vorschlag der EU-Kommission für eine Verordnung über In-vitro- Diagnostika am 26.09.2012 veröffentlicht Parallele Veröffentlichung des Vorschlages
Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203
 TRBS 1111 TRBS 2121 TRBS 1203") Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203 Achim Eckert 1/12 Am 3. Oktober 2002 ist die Betriebssicherheitsverordnung in Kraft getreten. Auch für den Gerüstbauer und den
Die Betriebssicherheitsverordnung (BetrSichV) TRBS 1111 TRBS 2121 TRBS 1203 Achim Eckert 1/12 Am 3. Oktober 2002 ist die Betriebssicherheitsverordnung in Kraft getreten. Auch für den Gerüstbauer und den
Baustellenverordnung. Verordnung über Sicherheit und Gesundheitsschutz auf Baustellen. Bestell-Nr.: BaustellV Gültig ab 1.
 ... q Verordnung über Sicherheit und Gesundheitsschutz auf Baustellen Baustellenverordnung Bestell-Nr.: BaustellV Gültig ab 1. Juli 1998 Achtung, diese Vorschrift kann nicht über die Süddeutsche Metall-Berufsgenossenschaft
... q Verordnung über Sicherheit und Gesundheitsschutz auf Baustellen Baustellenverordnung Bestell-Nr.: BaustellV Gültig ab 1. Juli 1998 Achtung, diese Vorschrift kann nicht über die Süddeutsche Metall-Berufsgenossenschaft
Workshop Fundraising, Spenden & Sponsoring. 16. Juni 2014 Dr. Robin Rumler Präsident
 Workshop Fundraising, Spenden & Sponsoring 16. Juni 2014 Dr. Robin Rumler Präsident Die Pharmig auf einen Blick Pharmig Verband der pharmazeutischen Industrie Österreichs seit 1954 freiwillige Interessensvertretung
Workshop Fundraising, Spenden & Sponsoring 16. Juni 2014 Dr. Robin Rumler Präsident Die Pharmig auf einen Blick Pharmig Verband der pharmazeutischen Industrie Österreichs seit 1954 freiwillige Interessensvertretung
Medizinprodukte im Handgepäck
 Medizinprodukte im Handgepäck Hannes Mühlenberg Consultant Medical Devices 2014 infoteam Software AG V 3 Seite 1 Motivation Medizinprodukte auf Reisen Grenzüberschreitende Nutzung von Medizinprodukten
Medizinprodukte im Handgepäck Hannes Mühlenberg Consultant Medical Devices 2014 infoteam Software AG V 3 Seite 1 Motivation Medizinprodukte auf Reisen Grenzüberschreitende Nutzung von Medizinprodukten
Verordnung, mit der die Gewebeentnahmeeinrichtungsverordnung geändert wird. Vorblatt. Ziele. Inhalt
 1 von 5 Verordnung, mit der die Gewebeentnahmeeinrichtungsverordnung geändert wird Einbringende Stelle: BMG Laufendes Finanzjahr: 2014 Inkrafttreten/ Wirksamwerden: 2014 Vorblatt Ziele - Erhöhung der Sicherheit
1 von 5 Verordnung, mit der die Gewebeentnahmeeinrichtungsverordnung geändert wird Einbringende Stelle: BMG Laufendes Finanzjahr: 2014 Inkrafttreten/ Wirksamwerden: 2014 Vorblatt Ziele - Erhöhung der Sicherheit
Die Sicht der EMEA. für Zulassungs-, Therapie- und. Berlin-Brandenburgische Akademie der
 Lebensqualität und Patientennutzen Die Sicht der EMEA PMS-Workshop: Lebensqualität als Kriterium für Zulassungs-, Therapie- und Erstattungsentscheidungen. Berlin-Brandenburgische Akademie der Wissenschaften
Lebensqualität und Patientennutzen Die Sicht der EMEA PMS-Workshop: Lebensqualität als Kriterium für Zulassungs-, Therapie- und Erstattungsentscheidungen. Berlin-Brandenburgische Akademie der Wissenschaften
Antrag zum Baumusterprüfverfahren (Konformitätsbewertungsverfahren)
") Arbeitsgebiet: Grundlagen Antrag zum Baumusterprüfverfahren (Konformitätsbewertungsverfahren) Akkreditierte Zertifizierungsstelle SCESp 008 Bestell-Nr. CE08-1.d Europäisch notifiziert, Kenn-Nr. 1246 Ausgabedatum
Arbeitsgebiet: Grundlagen Antrag zum Baumusterprüfverfahren (Konformitätsbewertungsverfahren) Akkreditierte Zertifizierungsstelle SCESp 008 Bestell-Nr. CE08-1.d Europäisch notifiziert, Kenn-Nr. 1246 Ausgabedatum
Gesundheits- und Medizin Apps: Stellen sie ein Sicherheitsrisiko dar?
 Gesundheits- und Medizin Apps: Stellen sie ein Sicherheitsrisiko dar? Apps für Smartphones werden immer populärer und erleichtern uns den schnellen Zugriff auf bestimmte Informationen. Seit ein paar Jahren
Gesundheits- und Medizin Apps: Stellen sie ein Sicherheitsrisiko dar? Apps für Smartphones werden immer populärer und erleichtern uns den schnellen Zugriff auf bestimmte Informationen. Seit ein paar Jahren
DAS NEUE GESETZ ÜBER FACTORING ( Amtsblatt der RS, Nr.62/2013)
") DAS NEUE GESETZ ÜBER FACTORING ( Amtsblatt der RS, Nr.62/2013) I Einleitung Das Parlament der Republik Serbien hat das Gesetz über Factoring verabschiedet, welches am 24. Juli 2013 in Kraft getreten ist.
DAS NEUE GESETZ ÜBER FACTORING ( Amtsblatt der RS, Nr.62/2013) I Einleitung Das Parlament der Republik Serbien hat das Gesetz über Factoring verabschiedet, welches am 24. Juli 2013 in Kraft getreten ist.
Umgang mit Explantaten
 Umgang mit Explantaten Aktuelle Entwicklung Implantate / Explantate Warum wird dieses Thema plötzlich so wichtig? Wem gehört das Explantat? Was muß berücksichtigt werden? Welche Aufgabe betrifft die ZSVA?
Umgang mit Explantaten Aktuelle Entwicklung Implantate / Explantate Warum wird dieses Thema plötzlich so wichtig? Wem gehört das Explantat? Was muß berücksichtigt werden? Welche Aufgabe betrifft die ZSVA?
Medizintechnik und IT
 Medizintechnik und IT Software als Medizinprodukt Alarmierung St.-Marien-Hospital Lünen 10.12.2013 1 MDD 2007/47/EG - Software Software als solche ist ein Medizinprodukt, wenn sie spezifisch vom Hersteller
Medizintechnik und IT Software als Medizinprodukt Alarmierung St.-Marien-Hospital Lünen 10.12.2013 1 MDD 2007/47/EG - Software Software als solche ist ein Medizinprodukt, wenn sie spezifisch vom Hersteller
Amtsblatt der Europäischen Gemeinschaften. (Veröffentlichungsbedürftige Rechtsakte) VERORDNUNG (EG) Nr. 150/2003 DES RATES vom 21.
 VERORDNUNG (EG) Nr. 150/2003 DES RATES vom 21.") 30.1.2003 L 25/1 I (Veröffentlichungsbedürftige Rechtsakte) VERORDNUNG (EG) Nr. 150/2003 S RATES vom 21. Januar 2003 zur Aussetzung der Einfuhrabgaben für bestimmte Waffen und militärische Ausrüstungsgüter
30.1.2003 L 25/1 I (Veröffentlichungsbedürftige Rechtsakte) VERORDNUNG (EG) Nr. 150/2003 S RATES vom 21. Januar 2003 zur Aussetzung der Einfuhrabgaben für bestimmte Waffen und militärische Ausrüstungsgüter
Qualitätsmanagement und Gesetzgebung
 Qualitätsmanagement und Gesetzgebung bei Medizinprodukten Gesetzliche Grundlagen und ddaraus resultierende Anforderungen für den Hersteller Unter Verwendung von Material von Dr. Johann Rader TÜV SÜD Product
Qualitätsmanagement und Gesetzgebung bei Medizinprodukten Gesetzliche Grundlagen und ddaraus resultierende Anforderungen für den Hersteller Unter Verwendung von Material von Dr. Johann Rader TÜV SÜD Product
Einteilung der Medizinprodukte nach RKI (Risikobewertung und Einstufung von Medizinprodukten vor der Aufbereitung)
") ÖGSV Fachkundelehrgang I 11 Einteilung der Medizinprodukte nach RKI (Risikobewertung und Einstufung von Medizinprodukten vor der Aufbereitung) M.T. Enko 2015 Inhalt 1 Ziel des Unterrichtes:... 3 2 Änderungen
ÖGSV Fachkundelehrgang I 11 Einteilung der Medizinprodukte nach RKI (Risikobewertung und Einstufung von Medizinprodukten vor der Aufbereitung) M.T. Enko 2015 Inhalt 1 Ziel des Unterrichtes:... 3 2 Änderungen
Klinische Bewertung & Marktbeobachtung
 Klinische Bewertung & Marktbeobachtung, Senior Regulatory Affairs Manager EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Sicherheits- und Leistungsanforderungen an ein Medizinprodukt
Klinische Bewertung & Marktbeobachtung, Senior Regulatory Affairs Manager EU: Medical Device Recast Es bleibt kaum ein Stein mehr auf dem anderen Sicherheits- und Leistungsanforderungen an ein Medizinprodukt
Wesentliche Inhalte des Geräte- und Produktsicherheitsgesetzes
 Wesentliche Inhalte des Geräte- und Produktsicherheitsgesetzes (GPSG) 16. März 2005 Tag der Arbeitssicherheit, Fellbach Rüdiger BGZ Gliederung Anforderungen an an Hersteller und Produkte Verbraucherschutz
Wesentliche Inhalte des Geräte- und Produktsicherheitsgesetzes (GPSG) 16. März 2005 Tag der Arbeitssicherheit, Fellbach Rüdiger BGZ Gliederung Anforderungen an an Hersteller und Produkte Verbraucherschutz
Widerrufsbelehrung der redcoon GmbH
 Widerrufsbelehrung der redcoon GmbH Stand: September 2015 www.redcoon.de Inhaltsverzeichnis Widerrufsbelehrung Verträge für die Lieferung von Waren Seite 3 Muster-Widerrufsformular Seite 5 Widerrufsbelehrung
Widerrufsbelehrung der redcoon GmbH Stand: September 2015 www.redcoon.de Inhaltsverzeichnis Widerrufsbelehrung Verträge für die Lieferung von Waren Seite 3 Muster-Widerrufsformular Seite 5 Widerrufsbelehrung
Zulassung nach MID (Measurement Instruments Directive)
") Anwender - I n f o MID-Zulassung H 00.01 / 12.08 Zulassung nach MID (Measurement Instruments Directive) Inhaltsverzeichnis 1. Hinweis 2. Gesetzesgrundlage 3. Inhalte 4. Zählerkennzeichnung/Zulassungszeichen
Anwender - I n f o MID-Zulassung H 00.01 / 12.08 Zulassung nach MID (Measurement Instruments Directive) Inhaltsverzeichnis 1. Hinweis 2. Gesetzesgrundlage 3. Inhalte 4. Zählerkennzeichnung/Zulassungszeichen
GEMA Gesellschaft für musikalische Aufführungs- und mechanische Vervielfältigungsrechte Berlin
 GEMA Gesellschaft für musikalische Aufführungs- und mechanische Vervielfältigungsrechte Berlin Vergütungssätze VR-W 2 für die Nutzung von Werken des GEMA-Repertoires in Websites mit Electronic Commerce
GEMA Gesellschaft für musikalische Aufführungs- und mechanische Vervielfältigungsrechte Berlin Vergütungssätze VR-W 2 für die Nutzung von Werken des GEMA-Repertoires in Websites mit Electronic Commerce
Aufbereitung von Medizinprodukten
 R K I - R I C H T L I N I E N Aufbereitung von Medizinprodukten Erläuterungen zu der Übersicht Nachfolgend veröffentlichen wir eine Übersicht zur Aufbereitung von Medizinprodukten der Gruppen semikritisch
R K I - R I C H T L I N I E N Aufbereitung von Medizinprodukten Erläuterungen zu der Übersicht Nachfolgend veröffentlichen wir eine Übersicht zur Aufbereitung von Medizinprodukten der Gruppen semikritisch