Investigator-Initiierte Studien Von der Idee bis zur Durchführung
|
|
|
- Steffen Lange
- vor 8 Jahren
- Abrufe
Transkript
1 1 Investigator-Initiierte Studien Von der Idee bis zur Durchführung Klinische Studienzentrale (CSC) Martina Beckmann Gesundheitsmanager

2 2 Definition von Ärzten/Wissenschaftlern initiierte klinische Prüfungen (IIT) Fehlen eines kommerziellen Sponsors (Pharmaindustrie) = nicht-kommerzielle Prüfung ( = finanzieller Förderer ) häufige Ziele: Prüfungen der Phase IV Verbesserung bekannter Therapieund Behandlungskonzepte (Therapieoptimierung), Test zugelassener AM in anderer Indikation o. an neuem Patientengut aber auch andere Studienphasen mit nicht zugelassenen AM

3 3 Erstberatung Art der Prüfung (AMG, MPG, NIS, Sonstiges), mono- oder multizentrisch, ggfs. BfS (Antragsstellung notwendig, wenn ja welche?) Vorerfahrungen mit Prüfungen im Prüfzentrum, des Studienteams (GCP-Kurs?) finanzielle Situation (wer ist finanzieller Förderer Verträge im CSC und ReFo gegenlesen lassen), Kostenkalkulation mögliche Kooperationen: Labor, Apotheke, Biostatistik interne Vereinbarungen Beantragung Sponsorschaft durch die Universität
 finanzielle Situation (wer ist finanzieller Förderer Verträge im CSC und ReFo gegenlesen lassen),")
4 4 Hilfe beim Abschluss der Versicherung Patienteninformation (Inhalt) Erstellung eines CRF geplantes SAE-Management Hinweis auf Monitoring Antragstellung EK und BfArM Hilfe bei der Registrierung nach Art der Prüfung EudraCT Modul 1/2 und

5 5 Vorstellung des Deutsches Registers Klinische Studien (DRKS) zur Studienregistrierung ( Ausgabe von Formblättern (z.b. Studienanmeldeformulare im CSC für Apotheke, Biostatistik im CSC) Abschluss eines Sponsor-Prüfer-Vertrages/ Prüfzentrumsverträgen bei IITs zwischen Universität (vertreten durch den Dekan) in Zusammenarbeit mit Refo und Stabsstelle Recht und Prüfer sowie beteiligten Zentren Absprache zu geplanten Folgeberatungen

6 6 Behördeneinreichung AMG 40 (1) [ ] Die klinische Prüfung eines Arzneimittels bei Menschen darf vom Sponsor nur begonnen werden, wenn die zuständige Ethik-Kommission diese nach Maßgabe des 42 Abs. 1 zustimmend bewertet und die zuständige Bundesoberbehörde diese nach Maßgabe des 42 Abs. genehmigt hat.

7 7 AMG 42 (2) [ ] Der Sponsor hat dabei alle Angaben und Unterlagen vorzulegen, die diese zur Bewertung benötigt, insbesondere die Ergebnisse der analytischen und der pharmakologischtoxikologischen Prüfung sowie den Prüfplan und die klinischen Angaben zum Arzneimittel einschließlich der Prüferinformation. Investigator Brochure = IB Investigational Medicinal Product Dossier, IMPD Fachinformation = Summary of Product Characteristics = SmPC

8 8 Zuständige Bundesoberbehörden für Klinischen Prüfungen von Arzneimittelstudien in Deutschland Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) Paul Ehrlich Institut (PEI) Zuständig für Sera, Impfstoffe, Gewebs- und Knochenmarkszubereitung Bundesamt für Strahlenschutz (BfS)

9 9 Einzureichende Unterlagen (GCP-V 7 Abs.2) Antragsformular welches über das Internet im Dialog dem EudraCT erzeugt werden kann (Modul 1) Die dem Antrag beizufügenden Unterlagen können in deutscher oder in englischer Sprache abgefasst sein, soweit im Folgenden nichts anderes bestimmt ist. Antrag und Unterlagen sind zusätzlich auf einem elektronischen Datenträger einzureichen.

10 10 Änderung der Homepage am European Clinical Trial Database (EudraCT) Erster Schritt ist die Beantragung einer EudraCT-Nr.


11 11

12 12

13 13

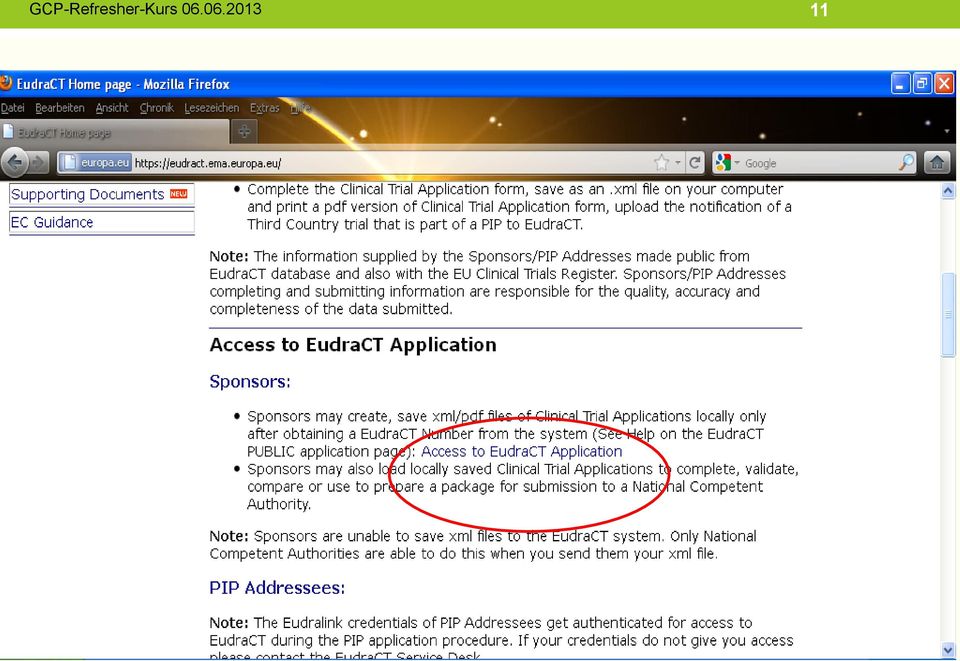
14 14 Meldeverfahren für die EudraCT Datenbank Schritt 1 Schritt 2 Schritt 3 Schritt 4 Beantragen eines Sicherheits-Codes Beantragen einer EudraCT- Studien-Nummer Ausfüllen des Clinical Trial Application Form Access to EudraCT Application Apply for Security Code Apply for EudraCT Number Click here to create a new Clinical Trial Application Bearbeiten eines gespeicherten Clinical Trial Application Form Click here to load a saved Clinical Trial Application Namens des Beantragenden eine -Adresse zum Empfang des Codes - Name der Organisation des Antragstellers - Sitz/Adresse (Land, Stadt) der Organisation des Antragstellers - ggf. durch den Sponsor vergebene Code-Nr. des Prüfplans - Name Antragsteller - Adresse - Antragsteller, zum Versand der EudraCT-Nummer Ausfüllen möglich lokal - im xml-format - gespeicherte Clinical Trial Application Form kann aufgerufen und geändert werden Der Sicherheitscode wird umgehend an die angegebene -Adresse zugestellt. Der Code gilt für die Beantragung einer klinischen Prüfung und ist nur 24 Stunden gültig. Die EudraCT-Nummer wird per zugestellt. Dieses ist unbedingt aufzubewahren. Es muss elektronisch und papierbasiert den Antragsunterlagen beigelegt werden. Das Clinical Trial Application Form muss vor und während des Ausfüllens regelmäßig lokal gespeichert werden. Es kann dann zur weiteren Bearbeitung wieder geladen werden. Das Clinical Trial Application Form muss, sowohl im xml- wie auch im pdf-format auf CD- ROM gespeichert und eingereicht werden.
 der")
15 15 Unterlagen Ethikkommission (GCP-V 7 Abs. 2) Die gleichen Unterlagen wie bei der BOB. Zusätzlich jedoch nach GCP-V 7 Abs. 3 folgende Unterlagen: Erläuterung der Bedeutung der klinischen Prüfung, Erklärung zur Einbeziehung möglicherweise vom Sponsor oder Prüfer abhängiger Personen, Angaben zur Finanzierung der klinischen Prüfung, Lebensläufe (wichtig nur Dienstanschrift) und andere geeignete Qualifikationsnachweise der Prüfer Angaben zu möglichen wirtschaftlichen und anderen Interessen der Prüfer im Zusammenhang mit den Prüfpräparaten

16 Anzeige der Teilnahme an einer Klinischen Prüfung bei der Landesoberbehörde nach Genehmigung durch EK und BOB 16 Landesverwaltungsamt Sachsen-Anhalt Referat Gesundheitswesen, Pharmazie Bereich 604.c - Pharmazie Ernst-Kamieth-Straße Halle/Saale Telefon ; Telefax pharmazie@lvwa.sachsen-anhalt.de Website: Dr. Bärbel zur Horst-Meyer (GMP-Inspektorin, GCP) baerbel.zurhorst-meyer@lvwa.sachsen-anhalt.de

17 17 ZLG-Formular: Formblatt zur Anzeige einer klinischen Prüfung gemäß 67 Abs. 1 und 3 AMG und 12 Abs. 1-3 GCP-V bei der zuständigen Landesbehörde Nur diese Anzeige, keine weiteren Prüfungsunterlagen Kann durch Prüfer oder Sponsor erfolgen Anzeigepflicht für beteiligte deutsche Prüfzentren bei der zuständigen Landesbehörde Ihres Bundeslandes; für internationale multizentrischen Prüfungen bei der zuständigen Bundesoberbehörde (competent authority) des Landes

18 18 GCP-V 10 (1) Nachträgliche Änderungen Änderungen einer von der zuständigen BOB genehmigten oder von der zuständigen EK zustimmend bewerteten klinischen Prüfung, die geeignet sind:

19 19 sich auf die Sicherheit der betroffenen Personen auszuwirken die Auslegung der wissenschaftlichen Dokumente, auf die die Prüfung gestützt wird, oder die wissenschaftliche Aussagekraft der Studienergebnisse zu beeinflussen, die Art der Leitung oder Durchführung der klinischen Prüfung wesentlich zu verändern, die Qualität oder Unbedenklichkeit der Prüfpräparate zu beeinträchtigen oder bei klinischen Prüfungen mit Arzneimitteln, die aus gentechnisch veränderten Organismen bestehen oder diese enthalten, die Risikobewertung für die Gesundheit nicht betroffener Personen und die Umwelt zu verändern

20 20 GCP-V 11 Maßnahmen zum Schutz vor unmittelbarer Gefahr (1) Unbeschadet des 10 treffen der Sponsor und der Prüfer unverzüglich alle gebotenen Maßnahmen zum Schutz der betroffenen Personen vor unmittelbarer Gefahr, wenn neue Umstände die Sicherheit der betroffenen Personen beeinträchtigen können.

21 21 GCP-V 13 (8) Der Sponsor unterrichtet die zuständige Behörde [ ] innerhalb von 90 Tagen über die Beendigung der klinischen Prüfung. Wurde die klinische Prüfung [ ] abgebrochen oder unterbrochen, erfolgt die Unterrichtung innerhalb von 15 Tagen
22 22 Die Registrierung einer klinischen Prüfung erfolgt nach der Erlangung aller Genehmigungen aber vor Einschluss des ersten Patienten. Registrierung in Primären Register soll verhindern, dass negative Studienergebnisse nicht publiziert werden Primäre Register sind von der WHO anerkannt und entsprechen bestimmten Qualitätskriterien.
 EU Clinical Trials Register (seit März 2011) Mit einer Registrierung im DRKS")
23 23 Es gibt vier von der WHO anerkannte primäre Studienregister: Clinicaltrials.gov Current Controlled Trial Deutsches Register Klinische Studien (seit Oktober 2008) EU Clinical Trials Register (seit März 2011) Mit einer Registrierung im DRKS sind damit die Anforderungen des ICMJE als Voraussetzung für eine Veröffentlichung erfüllt.
24 24 Damit eine Klinische Prüfung in allen internationalen und nationalen Registern wieder gefunden wird, etablierte die WHO eine Universal Trial Number (UTN) UTN (Universal Trial Number) eindeutigen Identifikation Klinischer Prüfungen sollte vom Sponsor oder Prüfer (Investigator) auf Homepage der WHO bezogen werden ist Teil der Identifikationsnummer der Klinischen Prüfung wird an alle Stellen, mit denen Daten ausgetauscht werden übermittelt sollte im Prüfplan aufgeführt werden UTN gilt weltweit als Identifikationsnummer Ist keine Registrierungsnummer, weshalb sie als Sekundär-ID zu übermitteln ist, bei Registrierung der Klinischen Prüfung
25 25
26 26
27 27 Definition Prüfplan GCP-V 3 (2) Beschreibung der Zielsetzung, Planung, Methodik, statistischen Erwägungen und Organisation einer klinischen Prüfung Definition Prüfplan ICH-GCP 1.44: A document that describes the objective(s), design, methodology, statistical considerations, and organization of a trial. The protocol usually also gives the background and rationale for the trial, but these could be provided in other protocol referenced documents. Abschnitt 6 widmet sich ausschließlich dem Prüfplan
28 Allgemeine Informationen 6.2. Hintergrundinformationen 6.3. Beschreibung der Zielkriterien und der Begründung für die Durchführung der klinischen Prüfungen 6.4. Studiendesign 6.5. Selections und Ausschlusskriterien für den Teilnehmer der Klinischen Prüfung
29 Intervention beim Teilnehmer der klinischen Prüfung 6.7 Wirksamkeitsparameter 6.8 Sicherheitsparameter 6.9 Statistik 6.10 Direkter Zugang zu den Source Data 6.11 Qualitätskontrolle und -sicherung 6.12 Ethische Aspekte
30 Datenmanagement und -speicherung 6.14 Finanzierung und Versicherung 6.15 Regelung zur Veröffentlichung 6.16 Supplements Da die Inhalte in ICH-GCP E6 knapp gehalten sind, Verweis auf: ICH-GCP E3 Structure and Content of Clinical Study Reports.
31 31 Der Prüfplan soll inhaltlich so formuliert sein, dass durch seine Befolgung die zwei Grundsätze von ICH-GCP erfüllt werden: Das die Rechte, Sicherheit und das Wohlergehen der Studienteilnehmer geschützt sind. Die Daten glaubwürdig sind.
32 32 Datenmanagement Hauptanforderungen nach ICH-GCP: - Entwickeln und Validieren von Prüfbögen (CRFs) - Validierung des EDV-Systems - SOPs (für die Nutzung des EDV-Systems) - Audit trail - Zugangskontrolle - Backup der Daten - Verblindung sicherstellen
33 33 Patienteneinwilligung = Informed Consent Form Anforderungen nach ICH-GCP : Unter der Seite finden Sie einen Assistenten zur Erstellung einer Patienteneinwilligung (PEW). Hier sind Erläuterung und Standardsätze vorzufinden, welche das Erstellen einer PEW erleichtern. Vorlagen beim Arbeitskreis der medizinischen Ethikkommissionen ak-med-ethik-komm.de/arbeitsunterlagen /Formulare
34 34 GCP-V 3 Prüfmedikation (3) Darreichungsformen von Wirkstoffen oder Placebos, die in einer klinischen Prüfung am Menschen getestet oder als Vergleichspräparate verwendet [ ] werden. GCP-V 4 (2) Der Sponsor muss sicherstellen, dass die Herstellung und Prüfung des Prüfpräparates den Angaben des [...] eingereichten Dossiers zum Prüfpräparat entspricht und [...]. Bei Verwendung zugelassener Arzneimittel [ ] gelten die Anforderungen als erfüllt, soweit der Sponsor bis auf die Kennzeichnung keine weiteren Herstellungsvorgänge unternimmt.
35 35 Rechtliche Grundlagen: ICH-GCP 4.6 Prüfarzt ICH-GCP Dokumentation ICH-GCP Kontrolle bzgl. der PM 5 (1) GCP-V Kennzeichnung der Prüfmedikation
36 36 Zusammenarbeit mit Apotheke!! Bestandsaufnahme PM Ausgabe der Prüfmedikation Rücknahme vom Patienten Patienten Compliance Prüfzentrum zum Sponsor ACHTUNG! Es kann mehr als ein Log geben am Zentrum: Ausgabe-Log pro Patient Ausgabe-Log für das Prüfzentrum Ausgabe-Log für jede PM
37 Arzneimittelsicherheit = Pharmakovigilanz 37 griech. Pharmacon = Heilmittel, Gift, Zaubermittel lat. Vigilantia = Wachheit, Schlauheit Bedeutet die laufende und systematische Überwachung der Sicherheit eines Fertigarzneimittels für Mensch oder Tier mit dem Ziel, dessen unerwünschte Wirkungen zu entdecken, zu beurteilen und zu verstehen, um entsprechende Maßnahmen zur Risikominimierung ergreifen zu können.
38 38 RL 2001/83/EG Änderungsrichtlinie 2010/84/EG RL 2001/20/EG zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln; vom Eudralex VOLUME 9A of The Rules Governing Medicinal Products in the European Union Guidelines on Pharmacovigilance for Medicinal Products for Human Use Aktuelle Fassung des AMG vom ICH-GCP E6 Leitlinie zur guten klinischen Praxis von 1997 Verordnung über die Anwendung der Guten Klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Arzneimitteln zur Anwendung am Menschen (GCP-Verordnung GCP-V)
39 39 RL 2010/84/EG = Änderungsrichtlinie zur RL 2001/83/EG RL zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel hinsichtlich Pharmakovigilanz RL 2001/20/EG Artikel 2: Begriffe m) unerwünschtes Ereignis n) Nebenwirkung o) Schwerwiegendes unerwünschtes Ereignis p) unerwartete Nebenwirkung Artikel 16: Berichte über Unerwünschte Ereignisse Artikel 17: Berichte über schwerwiegende Nebenwirkungen
40 40 AMG Begriffe (13) Nebenwirkungen Abschnitt 10: Pharmakovigilanz Bezieht sich im Großen und Ganzen auf den Inhaber der Zulassung! 63 AMG Meldeverpflichtungen für zugelassene AM
41 41 ICH-GCP E6 4. Prüfer 4.11 Meldung von unerwünschten Ereignissen 5. Sponsor 5.16 Informationen zur Sicherheit 5.17 Meldung von unerwünschten Arzneimittelwirkungen 6.8 Bewertung der Sicherheit ICH Guideline E2A: Clinical Safety Data Management: Definitions and Standards for Expedicted Reporting
42 42 3 Begriffsbestimmungen GCP-V (6) Unerwünschtes Ereignis (7) Nebenwirkung (8) Schwerwiegendes unerwünschtes Ereignis (9) Unerwartete Nebenwirkung 12 Anzeige-, Dokumentations- und Mitteilungspflichten des Prüfers 13 Dokumentations- und Mitteilungspflichten des Sponsors
43 43 Begriffe UE/AE Unerwünschtes Ereignis / Adverse Event SUE/SAE Schwerwiegendes UE / Serious AE UAW/ADR Unerwünschte Arzneimittelwirkung / Adverse Drug Reaction SAR Schwerwiegende Arzneimittelwirkung/ Serious Adverse Reaction SUSAR Suspected Unexpected Serious Adverse Reaction
44 44 Unerwünschtes Ereignis (UE), Adverse Event (AE) GCP-V 3 (6) Jedes nachteilige Vorkommnis, das einer betroffenen Person widerfährt, der ein Prüfpräparat verabreicht wurde, und das nicht notwendigerweise in ursächlichem Zusammenhang mit dieser Behandlung steht keine Kausalität ggf. anormale Laborbefunde neu aufgetretene/verstärkte Begleiterkrankungen (z. B. Erkältung, Durchfall, Migräne, Zahn-OP, Blasenentzündung) ggf. ausgeprägte Schwankungen in der Intensität von bekannten Begleiterkrankungen neue oder geänderte Begleitmedikation kann auf AEs hinweisen
45 45 Dokumentation Aes (im CRF + Line-Listing durch Prüfer) je Patient mit fortlaufender Nummer Beginn / Ende / Anhaltend (ongoing) Seriousness SAE Intensität (Schweregrad) Therapie erforderlich? Vorgehen bzgl. der Prüfmedikation
46 46 Schwerwiegendes Unerwünschtes Ereignis (SUE) / Serious Adverse Event (SAE) GCP-V 3 (8) Erfüllt ein AE eine der folgenden Bedingungen: tödlich oder lebensbedrohend erfordert oder verlängert eine stationäre Behandlung (nicht ein vorher geplanter Krankenhausaufenthalt) führt zu einer bleibenden- oder schwerwiegenden Behinderung führt zur Invalidität führt zu kongenitaler Anomalie oder einem Geburtsfehler
47 47 Mild Bewertung der Schweregrades / Intensity (Severity) Moderate Severe (schwer) Merke: Seriousness Severity Seriousness bewertet ob ein AE schwerwiegend ist Severity bewertet die Intensität eines AEs
48 48 Unerwünschte Arzneimittelwirkung (UAW) / Adverse Drug Reaction (ADR) AMG 4 (13), S.1 Nebenwirkungen sind bei Arzneimitteln, die zur Anwendung bei Menschen bestimmt sind, schädliche und unbeabsichtigte Reaktionen auf das Arzneimittel. GCP-V 3 (7) Nebenwirkung ist jede nachteilige Reaktion auf ein Prüfpräparat, unabhängig von der Dosierung. Die Kausalität muss bewertet und der ursächliche Zusammenhang gegeben sein Zweitbewerter (second assessment) In der Regel der LKP, sollte dieser aber der Melder des SAEs sein benötigt man wenigstens einen Stellvertreter (besser zwei oder mehr), damit fristgerechte Meldung von SAEs gewährleistet ist.
49 49 Achtung! Keine gesetzliche Vorgabe zur Definition von Kausalität! Kausalitätsbewertung der WHO ( certain (sicher) probable/likely (wahrscheinlich) possible (möglich) unlikely (unwahrscheinlich) conditional/unclassified (bedingt bewertbar) unassesable/unclassifiable (nicht bewertbar) Kausilitätsbewertung wird durch den Prüfarzt/Zweitbewerter und den Sponsor vorgenommen. Wird Kausalität nicht gleich bewertet, sind beide Einschätzungen zu berichten.
50 50 Bewertung, ob SAR erwartet (expected = SESAR) oder unerwartet (unexpected = SUSAR)) ist! Wird durch den Sponsor vorgenommen, mit Hilfe der relevanten Referenzdokumente: 1. Nicht zugelassenes AM: Investigator Brochure (IB) 2. Zugelassenes AM: Fachinformation = SmPC (Summary of Product Characteristics) SAR ist unerwartet und schwerwieged, wenn die Natur, der Schweregrad und die Folgen so nicht im Referenzdokument beschrieben sind!
51 51 Suspected Unexpected Serious Adverse Reaction (SUSAR) Schwerwiegend Unerwartet Kausalzusammenhang Definition beachten! Ca. 80% der an das BfArM gemeldeten SUSARs entsprechen nicht der Definition!
52 52 Expectedness SAR SUSAR Causality Seriousness SAE AE
53 Meldewege und pflichten 53 Prüfer. hat den Sponsor unverzüglich nach Kenntnisnahme über das Auftreten eines SAEs, ausgenommen Ereignisse über die laut Prüfplan oder Prüferinformation nicht unverzüglich berichtet werden muss, zu unterrichten und ihm anschließend einen ausführlichen schriftlichen Bericht zu übermitteln. Personenbezogene Daten sind vor ihrer Übermittlung unter Verwendung des Identifizierungscodes zu verblinden. 12 GCP-V
54 54

55 55
56 56
57 57 Sponsor hat bei jedem ihm bekannt gewordenen Verdachtsfall einer unerwarteten schwerwiegenden Nebenwirkung (SUSAR), die zu einem Todesfall geführt hat oder lebensbedrohlich ist, unverzüglich, spätestens aber innerhalb von sieben Tagen nach Bekanntwerden [...] und innerhalb von höchstens acht weiteren Tagen die weiteren relevanten Informationen zu übermitteln. 13 GCP-V
58 58.hat über jeden ihm bekannt gewordenen Verdachtsfall eines SUSAR unverzüglich, spätestens aber innerhalb von 15 Tagen nach Bekanntwerden, die zuständige Ethik-Kommission, die zuständige nationale Bundesoberbehörde, die Behörde [...] in deren Hoheitsgebiet (andere teilnehmende Länder) die klinische Prüfung auch durchgeführt wird, sowie die an der klinischen Prüfung beteiligten Prüfer zu unterrichten.
59 59 Meldung eines Verdachtsfall SUSARs an die Behörden muss immer entblindet erfolgen.
60 60 GCP-V 13 (4) Der Sponsor unterrichtet unverzüglich, spätestens aber innerhalb von 15 Tagen nach Bekanntwerden, die zuständige Bundesoberbehörde, die zuständige Ethik- Kommission und die zuständigen Behörden anderer Mitgliedstaaten [...] in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, über jeden Sachverhalt, der eine erneute Überprüfung der Nutzen-Risiko-Bewertung des Prüfpräparates erfordert.
61 61 Das gilt besonders für: 1. Einzelfallberichte von erwarteten schwerwiegenden Nebenwirkungen mit einem unerwarteten Ausgang, 2. eine Erhöhung der Häufigkeit erwarteter schwerwiegender Nebenwirkungen, die als klinisch relevant bewertet wird, 3. Verdachtsfälle schwerwiegender unerwarteter Nebenwirkungen, die sich ereigneten, nachdem die betroffene Person die klinische Prüfung bereits beendet hat, 4. Ereignisse im Zusammenhang mit der Studiendurchführung oder der Entwicklung des Prüfpräparates, die möglicherweise die Sicherheit der betroffenen Personen gefährden.
62 62 Der Sponsor hat alle ihm von den Prüfern mitgeteilten unerwünschten Ereignisse (SAEs) ausführlich zu dokumentieren (SAE-Line-Listing beim Sponsor). Diese Aufzeichnungen werden der zuständigen Bundesoberbehörde [...] in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, auf Anforderung übermittelt.
63 63 Development Safety Update Report (DSUR) ICH guideline E2F: Note for guidance on development safety update reports Achtung! Früher Annual Safety Report (ASR) Zusätzlich zu den Einzelfallmeldungen muss der Sponsor jährlich oder auf Verlangen einen Sicherheitsbericht über die klinische Prüfung erstellen Frist beginnt mit Datum der Genehmigung durch BOB = Date of Birth DSUR wird Substanzbezogen und nicht für einzelne Studie erstellt - aber häufig nur eine Studie mit dieser Substanz in der Sponsorverantwortung!!
64 64 Spezialfall Schwangerschaft Ist in der aktuellen gesetzlichen Regelung nicht ausreichend bedacht Meldung orientiert sich an Eudralex: Volume 9A (5.4) Meldung über das Auftreten und den Ausgang der Schwangerschaft Schwangerschaften gemäß AVB für klinische Prüfungen nicht mit versichert Meldung auch, wenn Partnerin von Prüfungsteilnehmer schwanger wird Meldung muss unverzüglich vom Prüfer an den Sponsor gehen Meldewege und -formulare gemäß Beschreibung im Prüfplan Führt meist zum Abbruch der Studie für Studienteilnehmerin
65 65 Data Monitoring Committee = DMC EMA: CHMP Guideline on Data Monitoring Committee, 2005 FDA: Guidance for Clinical Trial Sponsors - Establishment and Operation of Clinical Trial Data Monitoring Committees, 2006 DSMB = Data Safety Monitoring Board WHO: Operational Guideline for the Establishment and Functioning of Data and Safety Monitoring Committees = unabhängiges Kommittee das vom Sponsor (in Zusammenarbeit mit dem LKP) eingesetzt werden kann!
66 Wann sollte ein DMC eingerichtet werden? Betrachtung der Indikation Studienendpunkte Studienpopulation bisher bekannte Charakteristik Prüfprodukt (z.b. Langzeitstudien) viele bekannte Nebenwirkungen Kinder, geistig Behinderte) 66 Besetzung: erfahrener klinischer Mediziner zur betreffenden Indikation, ausreichend biostatistische Erfahrung Erfahrung bei ethischen Fragestellungen wenigstens der Vorsitzende sollte schon in einem DMC gearbeitet haben!
67 67 Aufgaben eines DMC Beratung des Sponsors Beobachtung des Fortgangs, Bewertung der Sicherheitsdaten (Zwischenanalysen), Anpassung der Risiko-Nutzen-Bewertung Empfehlungen für den Sponsor zu Fortgang, Unterbrechung, Abbruch / Beendigung (auch im positiven Sinne möglich) oder Änderung Vorgehen DMC sollte im Prüfplan oder Manual genau beschrieben werden Verantwortlichkeiten, Meldewege Fristen für Meetings Art (offen oder geschlossen) Regelung der Entblindung ist wichtig

68 68 Pharmakovigilanz bei NIS Im Beobachtungsplan wird der Berichtsprozess über beobachtete unerwünschte Nebenwirkungen von den Studienärzten an den Auftraggeber der NIS (z. B. Studienleiter, Zulasser oder Behörde) festgelegt. Ausfüllen des Berichtsbogen über unerwünschte Arzneimittelwirkungen nach 62 AMG und Weiterleitung an BfArM oder PEI, eventuell Arzneimittelkommission der deutschen Apotheker Vorgehen nach Routinevorgabe (als würde aus der täglichen Praxis heraus gemeldet werden)
69 69 Ergebnisbericht Verpflichtung zur Übermittlung von Ergebnisberichten klinischer Prüfungen gemäß 42b AMG
70 70
71 71 Quellen 1. alle relevanten Gesetze und Verordnungen 2. Internet-Seiten des BfArM, DRKS, EMA, ZLG, 3. Webinar Änderung des AMG Auswirkungen auf Klinische Prüfungen durch ZKS Köln am 17. Oktober 2012, Vortrag Dr. Andreas Stöhr GCP-Refresher-Kurs
Investigator-Initiierte Studien Von der Idee bis zur Durchführung
 1 Investigator-Initiierte Studien Von der Idee bis zur Durchführung Klinische Studienzentrale (CSC) Martina Beckmann Gesundheitsmanager 2 Übersicht Erstberatung Einreichung, Anzeige, Registrierung wesentliche
1 Investigator-Initiierte Studien Von der Idee bis zur Durchführung Klinische Studienzentrale (CSC) Martina Beckmann Gesundheitsmanager 2 Übersicht Erstberatung Einreichung, Anzeige, Registrierung wesentliche
Rahmenbedingungen bei der Durchführung klinischer Prüfungen (unter Berücksichtigung von IITs)
") Rahmenbedingungen bei der Durchführung klinischer Prüfungen (unter Berücksichtigung von IITs) Dr. Kay Stolzenburg Area Quality Consultant Lilly Deutschland GmbH Medizinische Abteilung Rahmenbedingungen
Rahmenbedingungen bei der Durchführung klinischer Prüfungen (unter Berücksichtigung von IITs) Dr. Kay Stolzenburg Area Quality Consultant Lilly Deutschland GmbH Medizinische Abteilung Rahmenbedingungen
Investigator-Initiierte Studien Von der Idee bis zur Durchführung
 1 Investigator-Initiierte Studien Von der Idee bis zur Durchführung Klinische Studienzentrale (CSC) Martina Beckmann Gesundheitsmanager 2 Definition von Ärzten/Wissenschaftlern initiierte klinische Prüfungen
1 Investigator-Initiierte Studien Von der Idee bis zur Durchführung Klinische Studienzentrale (CSC) Martina Beckmann Gesundheitsmanager 2 Definition von Ärzten/Wissenschaftlern initiierte klinische Prüfungen
Investigator Initiated Trials
 Investigator Initiated Trials Die Universität in der Rolle des Sponsors nach AMG - Rechtliche Aspekte - Alexander May, LL.M. Seite 1 Überblick Rechtliche Rahmenbedingungen 12. AMG-Novelle Der Sponsor und
Investigator Initiated Trials Die Universität in der Rolle des Sponsors nach AMG - Rechtliche Aspekte - Alexander May, LL.M. Seite 1 Überblick Rechtliche Rahmenbedingungen 12. AMG-Novelle Der Sponsor und
1. Hamburger Pharma Tag 17. April 2012. Pharmakovigilanzregelungen Ein wirkliches Plus für die Arzneimittelsicherheit?
 1. Hamburger Pharma Tag 17. April 2012 Pharmakovigilanzregelungen Ein wirkliches Plus für die Arzneimittelsicherheit? Simone Winnands Agenda Definitionen zur Pharmakovigilanz Pharmakovigilanz im Zulassungsumfeld
1. Hamburger Pharma Tag 17. April 2012 Pharmakovigilanzregelungen Ein wirkliches Plus für die Arzneimittelsicherheit? Simone Winnands Agenda Definitionen zur Pharmakovigilanz Pharmakovigilanz im Zulassungsumfeld
VERGLEICH. von NICHT-INTERVENTIONELLEN STUDIEN (NIS) mit KLINISCHEN PRÜFUNGEN (AMG) Klinische Studienzentrale (CSC UMMD ) Antje Wiede
 mit KLINISCHEN PRÜFUNGEN (AMG) Klinische Studienzentrale (CSC UMMD ) Antje Wiede") 1 VERGLEICH von NICHT-INTERVENTIONELLEN STUDIEN (NIS) mit KLINISCHEN PRÜFUNGEN (AMG) Klinische Studienzentrale (CSC UMMD ) Antje Wiede 2 Überblick Definitionen Ziele/Indikation einer NIS Begriff der Intervention
1 VERGLEICH von NICHT-INTERVENTIONELLEN STUDIEN (NIS) mit KLINISCHEN PRÜFUNGEN (AMG) Klinische Studienzentrale (CSC UMMD ) Antje Wiede 2 Überblick Definitionen Ziele/Indikation einer NIS Begriff der Intervention
Meldung von Serious Adverse Events (SAE) innerhalb von klinischen Studien
 innerhalb von klinischen Studien") Meldung von Serious Adverse Events (SAE) innerhalb von klinischen Studien Marga Lang-Welzenbach Strahlenklinik Warum dieses Thema beim Studientreffen? Ethikkommissionen empfehlen einen Qualifikationsnachweis
Meldung von Serious Adverse Events (SAE) innerhalb von klinischen Studien Marga Lang-Welzenbach Strahlenklinik Warum dieses Thema beim Studientreffen? Ethikkommissionen empfehlen einen Qualifikationsnachweis
Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach dem Arzneimittelgesetz (AMG) bei Investigator Initiated Trials (IITs)
 bei Investigator Initiated Trials (IITs)") 1 Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach dem Arzneimittelgesetz (AMG) bei Investigator Initiated Trials (IITs) Klinische Studienzentrale (CSC) Juliane Thapa 2 Inhaltsverzeichnis
1 Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach dem Arzneimittelgesetz (AMG) bei Investigator Initiated Trials (IITs) Klinische Studienzentrale (CSC) Juliane Thapa 2 Inhaltsverzeichnis
Angenommen am 14. April 2005
 05/DE WP 107 Arbeitsdokument Festlegung eines Kooperationsverfahrens zwecks Abgabe gemeinsamer Stellungnahmen zur Angemessenheit der verbindlich festgelegten unternehmensinternen Datenschutzgarantien Angenommen
05/DE WP 107 Arbeitsdokument Festlegung eines Kooperationsverfahrens zwecks Abgabe gemeinsamer Stellungnahmen zur Angemessenheit der verbindlich festgelegten unternehmensinternen Datenschutzgarantien Angenommen
Elektronische Unterstützung der Antragsstellung in Erasmus+ www.eu.daad.de/datenbanken
 Elektronische Unterstützung der Antragsstellung in Erasmus+ www.eu.daad.de/datenbanken 1 Schritte zum Mobilitätsantrag Beantragung der ECHE (Erasmus Charter for Higher Education 2014-2020) Registrierung
Elektronische Unterstützung der Antragsstellung in Erasmus+ www.eu.daad.de/datenbanken 1 Schritte zum Mobilitätsantrag Beantragung der ECHE (Erasmus Charter for Higher Education 2014-2020) Registrierung
Meldung der Waffennummern (Waffenkennzeichen) nach der Feuerwaffenverordnung der EU
 nach der Feuerwaffenverordnung der EU") Meldung der Waffennummern (Waffenkennzeichen) nach der Feuerwaffenverordnung der EU Meldung der Waffennummern (Waffenkennzeichen) 2 Allgemeine Hinweise Wenn Sie eine Nationale Ausfuhrgenehmigung oder eine
Meldung der Waffennummern (Waffenkennzeichen) nach der Feuerwaffenverordnung der EU Meldung der Waffennummern (Waffenkennzeichen) 2 Allgemeine Hinweise Wenn Sie eine Nationale Ausfuhrgenehmigung oder eine
Das BASG / AGES PharmMed
 CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
CMI-WORKSHOP Dipl.-Ing. Meinrad Guggenbichler Institut Inspektionen, Medizinprodukte und Hämovigilanz Das BASG / AGES PharmMed Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) ist die Aufsichtsbehörde
EMPFEHLUNG DER KOMMISSION. vom 13.1.2010
 EUROPÄISCHE KOMMISSION Brüssel, den 13.1.2010 K(2010)19 endgültig EMPFEHLUNG R KOMMISSION vom 13.1.2010 für den sicheren elektronischem Datenaustausch zwischen den Mitgliedstaaten zur Überprüfung der Einzigkeit
EUROPÄISCHE KOMMISSION Brüssel, den 13.1.2010 K(2010)19 endgültig EMPFEHLUNG R KOMMISSION vom 13.1.2010 für den sicheren elektronischem Datenaustausch zwischen den Mitgliedstaaten zur Überprüfung der Einzigkeit
Hinweise zum elektronischen Meldeformular
 Paul-Ehrlich-Institut Postfach 63207 Langen Jochen Halbauer Referat Pharmakovigilanz 2 Tel. +49 (0) 6103 77 3114 Fax +49 (0) 6103 77 1268 E-Mail pharmakovigilanz2@pei.de 22.06.2015 Hinweise zum elektronischen
Paul-Ehrlich-Institut Postfach 63207 Langen Jochen Halbauer Referat Pharmakovigilanz 2 Tel. +49 (0) 6103 77 3114 Fax +49 (0) 6103 77 1268 E-Mail pharmakovigilanz2@pei.de 22.06.2015 Hinweise zum elektronischen
IIT / AWB / NIS. Investigator Initiated Trials. Investigator Initiated Trials (IIT) 11.11.2015. IIT = Investigator Initiated Trials
 11.11.2015. IIT = Investigator Initiated Trials") IIT / AWB / NIS IIT = Investigator Initiated Trials AWB = Anwendungsbeobachtungen NIS = Nicht-interventionelle Studien Medical-Advisor-IIT-AWB-NIS 11.11.2015 Healthcare Marketing Dr. Umbach & Partner www.umbachpartner.com
IIT / AWB / NIS IIT = Investigator Initiated Trials AWB = Anwendungsbeobachtungen NIS = Nicht-interventionelle Studien Medical-Advisor-IIT-AWB-NIS 11.11.2015 Healthcare Marketing Dr. Umbach & Partner www.umbachpartner.com
Service-Angebote des BPI
 Pharmakovigilanz Service-Angebote des BPI BPI-Pharmakovigilanz Knowledge Base Mit dem EU-Pharmapackage und dem Zweiten Gesetz zur Änderung arzneimittelrechtlicher und anderer Vorschriften vom 19. Oktober
Pharmakovigilanz Service-Angebote des BPI BPI-Pharmakovigilanz Knowledge Base Mit dem EU-Pharmapackage und dem Zweiten Gesetz zur Änderung arzneimittelrechtlicher und anderer Vorschriften vom 19. Oktober
BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH. Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II
 1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
1 von 5 BUNDESGESETZBLATT FÜR DIE REPUBLIK ÖSTERREICH Jahrgang 2004 Ausgegeben am 28. Jänner 2004 Teil II 57. Verordnung: Konformitätsbewertung von Medizinprodukten [CELEX-Nr.: 32000L0070, 32001L0104,
Keine CE-Kennzeichnung ohne klinische Bewertung
 Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Seite 1 von 5 Keine CE-Kennzeichnung ohne klinische Bewertung Medizinprodukte können in der Regel nicht ohne klinische Daten und deren Bewertung auf den Markt gelangen. Zudem besteht für Medizinprodukte
Änderungen mit dem Humanforschungsgesetz Neue Vorlagen
 Departement Klinische Forschung Clinical Trial Unit Änderungen mit dem Humanforschungsgesetz Neue Vorlagen HFG News 3 Astrid Mattes 09. Januar 2014 -Seite 5 revidiert am 24.01.2014- Werkzeug zur Risikoklassifizierung
Departement Klinische Forschung Clinical Trial Unit Änderungen mit dem Humanforschungsgesetz Neue Vorlagen HFG News 3 Astrid Mattes 09. Januar 2014 -Seite 5 revidiert am 24.01.2014- Werkzeug zur Risikoklassifizierung
Registrierung von Abschlussprüfern aus Drittländern Formular A (DE)
") ABSCHLUSSPRÜFERAUFSICHTSKOMMISSION AUDITOROVERSIGHTC OMMISSION Registrierung von Abschlussprüfern aus Drittländern Formular A (DE) Formular zur Registrierung von Prüfungsunternehmen aus einem Drittland
ABSCHLUSSPRÜFERAUFSICHTSKOMMISSION AUDITOROVERSIGHTC OMMISSION Registrierung von Abschlussprüfern aus Drittländern Formular A (DE) Formular zur Registrierung von Prüfungsunternehmen aus einem Drittland
Maintenance & Re-Zertifizierung
 Zertifizierung nach Technischen Richtlinien Maintenance & Re-Zertifizierung Version 1.2 vom 15.06.2009 Bundesamt für Sicherheit in der Informationstechnik Postfach 20 03 63 53133 Bonn Tel.: +49 22899 9582-0
Zertifizierung nach Technischen Richtlinien Maintenance & Re-Zertifizierung Version 1.2 vom 15.06.2009 Bundesamt für Sicherheit in der Informationstechnik Postfach 20 03 63 53133 Bonn Tel.: +49 22899 9582-0
28.8.2009 Amtsblatt der Europäischen Union L 226/3
 28.8.2009 Amtsblatt der Europäischen Union L 226/3 VERORDNUNG (EG) Nr. 780/2009 DER KOMMISSION vom 27. August 2009 zur Festlegung der Durchführungsbestimmungen zu Artikel 28a Absatz 2 Unterabsatz 3 sowie
28.8.2009 Amtsblatt der Europäischen Union L 226/3 VERORDNUNG (EG) Nr. 780/2009 DER KOMMISSION vom 27. August 2009 zur Festlegung der Durchführungsbestimmungen zu Artikel 28a Absatz 2 Unterabsatz 3 sowie
PRÜFMODUL D UND CD. 1 Zweck. 2 Durchführung. 2.1 Allgemeines. 2.2 Antrag
 1 Zweck PRÜFMODUL D UND CD Diese Anweisung dient als Basis für unsere Kunden zur Information des Ablaufes der folgenden EG-Prüfung nach folgenden Prüfmodulen: D CD Es beschreibt die Aufgabe der benannten
1 Zweck PRÜFMODUL D UND CD Diese Anweisung dient als Basis für unsere Kunden zur Information des Ablaufes der folgenden EG-Prüfung nach folgenden Prüfmodulen: D CD Es beschreibt die Aufgabe der benannten
Aufgaben der MP-Berater und Sicherheitsbeauftragten im Medizinprodukte-Beobachtungs- und Meldesystem
 Aufgaben der MP-Berater und Sicherheitsbeauftragten im Medizinprodukte-Beobachtungs- und Meldesystem Referent: Ralf Kitschmann Seite 1/ 09-2009 30 MPG*, Sicherheitsbeauftragter für Medizinprodukte * Achtung:
Aufgaben der MP-Berater und Sicherheitsbeauftragten im Medizinprodukte-Beobachtungs- und Meldesystem Referent: Ralf Kitschmann Seite 1/ 09-2009 30 MPG*, Sicherheitsbeauftragter für Medizinprodukte * Achtung:
Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach AMG (IIT)
") 1 Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach AMG (IIT) Klinische Studienzentrale (CSC) Juliane Thapa 2 Inhaltsverzeichnis Magdeburg 3 AN DER KLINISCHEN PRÜFUNG BETEILIGTE PERSONEN
1 Die Rolle des Sponsors bei der Durchführung Klinischer Prüfungen nach AMG (IIT) Klinische Studienzentrale (CSC) Juliane Thapa 2 Inhaltsverzeichnis Magdeburg 3 AN DER KLINISCHEN PRÜFUNG BETEILIGTE PERSONEN
Klinische Prüfungen mit Medizinprodukten
 1 Klinische Prüfungen mit Medizinprodukten Klinische Studienzentrale (CSC) Antje Wiede 2 Überblick Begriffe relevante Regularien und Gesetze GCP bei KP mit Medizinprodukten Antragstellung und Genehmigung
1 Klinische Prüfungen mit Medizinprodukten Klinische Studienzentrale (CSC) Antje Wiede 2 Überblick Begriffe relevante Regularien und Gesetze GCP bei KP mit Medizinprodukten Antragstellung und Genehmigung
Hinweise zum elektronischen Meldeformular
 BASG / AGES Institut Überwachung Traisengasse 5, 1200 Wien, Österreich Hinweise zum elektronischen Meldeformular Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) hat gemeinsam mit dem BfArM ein
BASG / AGES Institut Überwachung Traisengasse 5, 1200 Wien, Österreich Hinweise zum elektronischen Meldeformular Das Bundesamt für Sicherheit im Gesundheitswesen (BASG) hat gemeinsam mit dem BfArM ein
!"#$%&'()*& +,-&',.-)/,.012-34151"/46!1
*& +,-&',.-)/,.012-34151/46!1") !"#$%&'()*& +,-&',.-)/,.012-34151"/46!1 111111111111111111111111111111111111117/8'1#9.:&1+$+;1"0),)*.01
!"#$%&'()*& +,-&',.-)/,.012-34151"/46!1 111111111111111111111111111111111111117/8'1#9.:&1+$+;1"0),)*.01
Klinische Prüfungen von Arzneimitteln aktuelle Anforderungen
 Klinische Prüfungen von Arzneimitteln aktuelle Anforderungen Aus der Sicht der Überwachungsbehörde Sabine Hofsäss (Regierungspräsidium Karlsruhe) Gemeinsame Ziele: Sicherheit, Rechte und Wohl der Patienten
Klinische Prüfungen von Arzneimitteln aktuelle Anforderungen Aus der Sicht der Überwachungsbehörde Sabine Hofsäss (Regierungspräsidium Karlsruhe) Gemeinsame Ziele: Sicherheit, Rechte und Wohl der Patienten
WAS finde ich WO im Beipackzettel
 WAS finde ich WO im Beipackzettel Sie haben eine Frage zu Ihrem? Meist finden Sie die Antwort im Beipackzettel (offiziell "Gebrauchsinformation" genannt). Der Aufbau der Beipackzettel ist von den Behörden
WAS finde ich WO im Beipackzettel Sie haben eine Frage zu Ihrem? Meist finden Sie die Antwort im Beipackzettel (offiziell "Gebrauchsinformation" genannt). Der Aufbau der Beipackzettel ist von den Behörden
WS 2009/2010 Vorlesung - Spezielles Arzneimittelrecht. Mittwoch, 13. Januar 2010. Pandemie - Teil X
 Ministerium für,,, und WS 2009/2010 Vorlesung - Spezielles Arzneimittelrecht Mittwoch, 13. Januar 2010 Pandemie - Teil X -Dr. Michael Cramer-2010-01-13 Folie 1 Ministerium für,,, und Klinische Prüfungen
Ministerium für,,, und WS 2009/2010 Vorlesung - Spezielles Arzneimittelrecht Mittwoch, 13. Januar 2010 Pandemie - Teil X -Dr. Michael Cramer-2010-01-13 Folie 1 Ministerium für,,, und Klinische Prüfungen
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) Vom 20. Dezember 2001, BGBl. I S. 3854 geändert am 4. Dezember 2002, BGBl I S. 4456 zuletzt geändert am 13. Februar 2004, BGBl I S. 216
EU-Verordnung Nr. 1907/2006 (REACH)
") Was bedeutet REACH? REACH ist die EG-Verordnung Nr. 1907/2006 zur Registration, Evaluation, Authorization and Restriction of CHemicals (Registrierung, Bewertung und Zulassung von Chemikalien). Mit dieser
Was bedeutet REACH? REACH ist die EG-Verordnung Nr. 1907/2006 zur Registration, Evaluation, Authorization and Restriction of CHemicals (Registrierung, Bewertung und Zulassung von Chemikalien). Mit dieser
Anleitung öffentlicher Zugang einrichten
 TRK-DashBoard Anleitung öffentlicher Zugang einrichten Manual für Kunden VERSION DATUM AUTOR DATEINAME 1.0 8. SEPTEMBER 2011 HRR ANLEITUNG_OEFFENTLICHER_ZUGANG_DASHBOARD_V10 INHALT 1 ALLGEMEINE INFORMATIONEN...
TRK-DashBoard Anleitung öffentlicher Zugang einrichten Manual für Kunden VERSION DATUM AUTOR DATEINAME 1.0 8. SEPTEMBER 2011 HRR ANLEITUNG_OEFFENTLICHER_ZUGANG_DASHBOARD_V10 INHALT 1 ALLGEMEINE INFORMATIONEN...
Monitoring. Lucia Kacina. CTU Bern
 Monitoring Lucia Kacina CTU Bern Themen Einverständniserklärung Protokoll & Dokumentation (e)crf-einträge: Dos & Don ts Monitoringvisiten: Teilnehmer & Ablauf Monitoring: Aufgaben des Zentrums Monitoring
Monitoring Lucia Kacina CTU Bern Themen Einverständniserklärung Protokoll & Dokumentation (e)crf-einträge: Dos & Don ts Monitoringvisiten: Teilnehmer & Ablauf Monitoring: Aufgaben des Zentrums Monitoring
Richtlinie. des Gemeinsamen Bundesausschusses. zur Umsetzung der Regelungen in 62 für schwerwiegend chronisch Erkrankte ( Chroniker-Richtlinie )
") Richtlinie des Gemeinsamen Bundesausschusses zur Umsetzung der Regelungen in 62 für schwerwiegend chronisch Erkrankte ( Chroniker-Richtlinie ) in der Fassung vom 22. Januar 2004 veröffentlicht im Bundesanzeiger
Richtlinie des Gemeinsamen Bundesausschusses zur Umsetzung der Regelungen in 62 für schwerwiegend chronisch Erkrankte ( Chroniker-Richtlinie ) in der Fassung vom 22. Januar 2004 veröffentlicht im Bundesanzeiger
InVo. Information zu Verordnungen in der GKV. Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen. Stand: Februar 2010
 Nr. 1 2010 InVo Information zu Verordnungen in der GKV Stand: Februar 2010 Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen Bisher konnten Sie als Arzt Arzneimittel (z. B.
Nr. 1 2010 InVo Information zu Verordnungen in der GKV Stand: Februar 2010 Herstellung von Arzneimitteln durch Ärzte Anzeigepflicht bei Bezirksregierungen Bisher konnten Sie als Arzt Arzneimittel (z. B.
Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren
 Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit 1 Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren Politische Ebene Bundesbehörden Ministerien, insbesondere
Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit 1 Verteilung der Verantwortlichkeiten zuvor, Anzeigeverfahren Politische Ebene Bundesbehörden Ministerien, insbesondere
Besonderheiten zum Zulassungsverfahren von pathogeninaktivierten Blutkomponenten. W. Schwarz Paul-Ehrlich-Institut Langen
 Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel www.pei.de Besonderheiten zum Zulassungsverfahren von pathogeninaktivierten W. Schwarz Paul-Ehrlich-Institut Langen Zuerst verfolgte Vorgehensweise:
Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel www.pei.de Besonderheiten zum Zulassungsverfahren von pathogeninaktivierten W. Schwarz Paul-Ehrlich-Institut Langen Zuerst verfolgte Vorgehensweise:
Schritte zum Systempartner Stufe Großhandel
 Schritte zum Systempartner Stufe Großhandel Schritt 1: Anmeldung in der Software-Plattform https://qs-platform.info/ Falls Sie bislang noch kein QS- Systempartner sind, gehen Sie bitte auf die Startseite
Schritte zum Systempartner Stufe Großhandel Schritt 1: Anmeldung in der Software-Plattform https://qs-platform.info/ Falls Sie bislang noch kein QS- Systempartner sind, gehen Sie bitte auf die Startseite
Häufig wiederkehrende Fragen zur mündlichen Ergänzungsprüfung im Einzelnen:
 Mündliche Ergänzungsprüfung bei gewerblich-technischen und kaufmännischen Ausbildungsordnungen bis zum 31.12.2006 und für alle Ausbildungsordnungen ab 01.01.2007 Am 13. Dezember 2006 verabschiedete der
Mündliche Ergänzungsprüfung bei gewerblich-technischen und kaufmännischen Ausbildungsordnungen bis zum 31.12.2006 und für alle Ausbildungsordnungen ab 01.01.2007 Am 13. Dezember 2006 verabschiedete der
Gemeinsame Ausführungsordnung zum Madrider Abkommen 1 über die internationale Registrierung von Marken und zum Protokoll 2 zu diesem Abkommen
 BGBl. III - Ausgegeben am 9. März 2015 - Nr. 32 1 von 7 (Übersetzung) Gemeinsame Ausführungsordnung zum Madrider Abkommen 1 über die internationale Registrierung von Marken und zum Protokoll 2 zu diesem
BGBl. III - Ausgegeben am 9. März 2015 - Nr. 32 1 von 7 (Übersetzung) Gemeinsame Ausführungsordnung zum Madrider Abkommen 1 über die internationale Registrierung von Marken und zum Protokoll 2 zu diesem
Die Beschreibung bezieht sich auf die Version Dreamweaver 4.0. In der Version MX ist die Sitedefinition leicht geändert worden.
 In einer Website haben Seiten oft das gleiche Layout. Speziell beim Einsatz von Tabellen, in denen die Navigation auf der linken oder rechten Seite, oben oder unten eingesetzt wird. Diese Anteile der Website
In einer Website haben Seiten oft das gleiche Layout. Speziell beim Einsatz von Tabellen, in denen die Navigation auf der linken oder rechten Seite, oben oder unten eingesetzt wird. Diese Anteile der Website
Evaluation eines stichprobenartigen Monitorings bei Therapieoptimierungsstudien Ulrike Zettelmeyer
 internet: www.lymphome.de email: lymphome@medizin.uni-koeln.de Evaluation eines stichprobenartigen Monitorings bei Therapieoptimierungsstudien Ulrike Zettelmeyer Gliederung Deutsche Hodgkin Studiengruppe
internet: www.lymphome.de email: lymphome@medizin.uni-koeln.de Evaluation eines stichprobenartigen Monitorings bei Therapieoptimierungsstudien Ulrike Zettelmeyer Gliederung Deutsche Hodgkin Studiengruppe
Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV)
") 05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
05.07.2005 Verordnung über Medizinprodukte (Medizinprodukte-Verordnung - MPV) vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 13. Februar 2004 (BGBl. I S. 216)
Dopaminerge Substanzen und Impulskontrollstörungen
 - 2 - Bundesinstitut für Arzneimittel und Medizinprodukte BfArM Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Pharmazeutische Unternehmer (s. Anlage) Nachrichtlich: Stufenplanbeteiligte Postanschrift: Kurt-Georg-Kiesinger-Allee
- 2 - Bundesinstitut für Arzneimittel und Medizinprodukte BfArM Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Pharmazeutische Unternehmer (s. Anlage) Nachrichtlich: Stufenplanbeteiligte Postanschrift: Kurt-Georg-Kiesinger-Allee
nach 20 SGB IX" ( 3 der Vereinbarung zum internen Qualitätsmanagement nach 20 Abs. 2a SGB IX).
.") Information zum Verfahren zur Anerkennung von rehabilitationsspezifischen Qualitätsmanagement- Verfahren auf Ebene der BAR (gemäß 4 der Vereinbarung zum internen Qualitätsmanagement nach 20 Abs. 2a SGB
Information zum Verfahren zur Anerkennung von rehabilitationsspezifischen Qualitätsmanagement- Verfahren auf Ebene der BAR (gemäß 4 der Vereinbarung zum internen Qualitätsmanagement nach 20 Abs. 2a SGB
Wenn Sie kein in seinen Rechten verletzter Inhaber von Schutzrechten sind, melden Sie rechteverletzende Artikel bitte unserem Sicherheitsteam:
 Sehr geehrte Damen und Herren, vielen Dank, dass Sie sich um die Entfernung vermutlich rechteverletzender Angebote von unserem Marktplatz bemühen. Sollten Sie Inhaber gewerblicher Schutzrechte (z.b. Patente,
Sehr geehrte Damen und Herren, vielen Dank, dass Sie sich um die Entfernung vermutlich rechteverletzender Angebote von unserem Marktplatz bemühen. Sollten Sie Inhaber gewerblicher Schutzrechte (z.b. Patente,
Inhaltsübersicht Produktinformationsblatt zur Jahres-Reiserücktritts-Versicherung der Europäische Reiseversicherung AG
 Inhaltsübersicht Produktinformationsblatt zur Jahres-Reiserücktritts-Versicherung der Europäische Reiseversicherung AG 1. Produktinformationsblatt zur Jahres-Reiserücktritts-Versicherung mit Selbstbeteiligung
Inhaltsübersicht Produktinformationsblatt zur Jahres-Reiserücktritts-Versicherung der Europäische Reiseversicherung AG 1. Produktinformationsblatt zur Jahres-Reiserücktritts-Versicherung mit Selbstbeteiligung
BESCHLUSS DES GEMEINSAMEN EWR-AUSSCHUSSES Nr. 15/2001 vom 28. Februar 2001. zur Änderung des Anhangs IX (Finanzdienstleistungen) des EWR-Abkommens
 des EWR-Abkommens") BESCHLUSS DES GEMEINSAMEN EWR-AUSSCHUSSES Nr. 15/2001 vom 28. Februar 2001 zur Änderung des Anhangs IX (Finanzdienstleistungen) des EWR-Abkommens DER GEMEINSAME EWR-AUSSCHUSS - gestützt auf das Abkommen
BESCHLUSS DES GEMEINSAMEN EWR-AUSSCHUSSES Nr. 15/2001 vom 28. Februar 2001 zur Änderung des Anhangs IX (Finanzdienstleistungen) des EWR-Abkommens DER GEMEINSAME EWR-AUSSCHUSS - gestützt auf das Abkommen
Wir empfehlen Ihnen eine zeitnahe Bewerbung, da in jedem Halbjahr nur eine limitierte Anzahl an Bündnissen bewilligt werden können.
 Ich bin ein LeseHeld Bewerbungsformular zur Teilnahme am Leselernförderprojekt des Borromäusverein e.v. im Rahmen des Programms Kultur macht stark. Bündnisse für Bildung des Bundesministeriums für Bildung
Ich bin ein LeseHeld Bewerbungsformular zur Teilnahme am Leselernförderprojekt des Borromäusverein e.v. im Rahmen des Programms Kultur macht stark. Bündnisse für Bildung des Bundesministeriums für Bildung
Registrierung am Elterninformationssysytem: ClaXss Infoline
 elektronisches ElternInformationsSystem (EIS) Klicken Sie auf das Logo oder geben Sie in Ihrem Browser folgende Adresse ein: https://kommunalersprien.schule-eltern.info/infoline/claxss Diese Anleitung
elektronisches ElternInformationsSystem (EIS) Klicken Sie auf das Logo oder geben Sie in Ihrem Browser folgende Adresse ein: https://kommunalersprien.schule-eltern.info/infoline/claxss Diese Anleitung
Information und Beratung des Patienten bei der Abgabe von Arzneimitteln Erst- und Wiederholungsverordnung
 Leitlinie Kommentar Arbeitshilfe Leitlinie der Bundesapothekerkammer zur Qualitätssicherung Information und Beratung des Patienten bei der Abgabe von Arzneimitteln Erst- und Stand der Revision: 13.11.2013
Leitlinie Kommentar Arbeitshilfe Leitlinie der Bundesapothekerkammer zur Qualitätssicherung Information und Beratung des Patienten bei der Abgabe von Arzneimitteln Erst- und Stand der Revision: 13.11.2013
GCP-Schulung für Prüfer/Stellvertreter/Mitglieder der Prüfgruppe für klinische Prüfungen nach AMG
 GCP-Schulung für Prüfer/Stellvertreter/Mitglieder der Prüfgruppe für klinische Prüfungen nach AMG Kursleiter: Prof. Dr. Richard F. Schlenk Datum: 2. Schulungstag: 15.10.2014 Name Vorname Anleitung Tragen
GCP-Schulung für Prüfer/Stellvertreter/Mitglieder der Prüfgruppe für klinische Prüfungen nach AMG Kursleiter: Prof. Dr. Richard F. Schlenk Datum: 2. Schulungstag: 15.10.2014 Name Vorname Anleitung Tragen
Seite 1 von 14. Cookie-Einstellungen verschiedener Browser
 Seite 1 von 14 Cookie-Einstellungen verschiedener Browser Cookie-Einstellungen verschiedener Browser, 7. Dezember 2015 Inhaltsverzeichnis 1.Aktivierung von Cookies... 3 2.Cookies... 3 2.1.Wofu r braucht
Seite 1 von 14 Cookie-Einstellungen verschiedener Browser Cookie-Einstellungen verschiedener Browser, 7. Dezember 2015 Inhaltsverzeichnis 1.Aktivierung von Cookies... 3 2.Cookies... 3 2.1.Wofu r braucht
40-Tage-Wunder- Kurs. Umarme, was Du nicht ändern kannst.
 40-Tage-Wunder- Kurs Umarme, was Du nicht ändern kannst. Das sagt Wikipedia: Als Wunder (griechisch thauma) gilt umgangssprachlich ein Ereignis, dessen Zustandekommen man sich nicht erklären kann, so dass
40-Tage-Wunder- Kurs Umarme, was Du nicht ändern kannst. Das sagt Wikipedia: Als Wunder (griechisch thauma) gilt umgangssprachlich ein Ereignis, dessen Zustandekommen man sich nicht erklären kann, so dass
Dopaminerge Substanzen und Impulskontrollstörungen
 - 2 - Bundesinstitut für Arzneimittel und Medizinprodukte BfArM Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Pharmazeutische Unternehmer (s. Anlage) Nachrichtlich: Stufenplanbeteiligte Postanschrift: Kurt-Georg-Kiesinger-Allee
- 2 - Bundesinstitut für Arzneimittel und Medizinprodukte BfArM Kurt-Georg-Kiesinger-Allee 3 D-53175 Bonn Pharmazeutische Unternehmer (s. Anlage) Nachrichtlich: Stufenplanbeteiligte Postanschrift: Kurt-Georg-Kiesinger-Allee
CoCStom. Inhalt. 1. Monitoring und Query-Prozess. 2. SAE-Meldeweg. 3. Vorstellung des Investigator Site File (ISF) 21./22.11.
 21./22.11.") Prospective randomized multicentre investigator initiated study: Randomised trial comparing completeness of adjuvant chemotherapy after early versus late diverting stoma closure in low anterior resection
Prospective randomized multicentre investigator initiated study: Randomised trial comparing completeness of adjuvant chemotherapy after early versus late diverting stoma closure in low anterior resection
Patienteninformationsbroschüre Valproat
 Patienteninformationsbroschüre Valproat Informationen für Patientinnen Die Informationen in dieser Broschüre sind für Frauen bestimmt, denen Valproat verschrieben wird und die schwanger werden können (Frauen
Patienteninformationsbroschüre Valproat Informationen für Patientinnen Die Informationen in dieser Broschüre sind für Frauen bestimmt, denen Valproat verschrieben wird und die schwanger werden können (Frauen
Tevalo Handbuch v 1.1 vom 10.11.2011
 Tevalo Handbuch v 1.1 vom 10.11.2011 Inhalt Registrierung... 3 Kennwort vergessen... 3 Startseite nach dem Login... 4 Umfrage erstellen... 4 Fragebogen Vorschau... 7 Umfrage fertigstellen... 7 Öffentliche
Tevalo Handbuch v 1.1 vom 10.11.2011 Inhalt Registrierung... 3 Kennwort vergessen... 3 Startseite nach dem Login... 4 Umfrage erstellen... 4 Fragebogen Vorschau... 7 Umfrage fertigstellen... 7 Öffentliche
DAS GRÜNE REZEPT. Für eine sichere Medikation mit rezeptfreien Arzneimitteln
 DAS GRÜNE REZEPT Für eine sichere Medikation mit rezeptfreien Arzneimitteln Was ist das Grüne Rezept? Obwohl das Grüne Rezept schon seit Jahren in Arztpraxen verwendet wird, ist es vielen Patienten und
DAS GRÜNE REZEPT Für eine sichere Medikation mit rezeptfreien Arzneimitteln Was ist das Grüne Rezept? Obwohl das Grüne Rezept schon seit Jahren in Arztpraxen verwendet wird, ist es vielen Patienten und
Das Persönliche Budget in verständlicher Sprache
 Das Persönliche Budget in verständlicher Sprache Das Persönliche Budget mehr Selbstbestimmung, mehr Selbstständigkeit, mehr Selbstbewusstsein! Dieser Text soll den behinderten Menschen in Westfalen-Lippe,
Das Persönliche Budget in verständlicher Sprache Das Persönliche Budget mehr Selbstbestimmung, mehr Selbstständigkeit, mehr Selbstbewusstsein! Dieser Text soll den behinderten Menschen in Westfalen-Lippe,
Änderungen der MPBetreibV 2014
 Änderungen der MPBetreibV 2014 3 Instandhaltung von Medizinprodukten (1) Die Instandhaltung von Medizinprodukten umfasst insbesondere Instandhaltungsmaßnahmen und die Instandsetzung. Instandhaltungsmaßnahmen
Änderungen der MPBetreibV 2014 3 Instandhaltung von Medizinprodukten (1) Die Instandhaltung von Medizinprodukten umfasst insbesondere Instandhaltungsmaßnahmen und die Instandsetzung. Instandhaltungsmaßnahmen
Abweichungen. Anforderungen / Zitate aus den Rechtsvorschriften
 Abweichungen Anforderungen / Zitate aus den Rechtsvorschriften AMWHV [...] Alle Abweichungen im Prozess und von der Festlegung der Spezifikation sind zu dokumentieren und gründlich zu untersuchen. [...]
Abweichungen Anforderungen / Zitate aus den Rechtsvorschriften AMWHV [...] Alle Abweichungen im Prozess und von der Festlegung der Spezifikation sind zu dokumentieren und gründlich zu untersuchen. [...]
Antrag für ein Schlichtungs-Verfahren
 Eingangsstempel Antrag für ein Schlichtungs-Verfahren Dieser Antrag ist in Leichter Sprache geschrieben. Das sieht man auch am gelben, runden Zeichen. Im Text finden Sie immer wieder unterstrichene Wörter.
Eingangsstempel Antrag für ein Schlichtungs-Verfahren Dieser Antrag ist in Leichter Sprache geschrieben. Das sieht man auch am gelben, runden Zeichen. Im Text finden Sie immer wieder unterstrichene Wörter.
Abschnitt 1 Anwendungsbereich und Allgemeine Anforderungen an die Konformitätsbewertung 1 Anwendungsbereich
 13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
13.06.2007 Verordnung über Medizinprodukte - (Medizinprodukte-Verordnung - MPV)* vom 20. Dezember 2001 (BGBl. I S. 3854), zuletzt geändert durch Artikel 1 der Verordnung vom 16. Februar 2007 (BGBl. I S.
16. AMG Novelle Neuerungen für die Klinische Prüfung
 16. AMG Novelle Neuerungen für die Klinische Prüfung Dr. med. Kerstin Breithaupt-Grögler Fachärztin für klinische Pharmakologie Frankfurt am Main e-mail: breithaupt-groegler@t-online.de -kbr-2013 16. AMG
16. AMG Novelle Neuerungen für die Klinische Prüfung Dr. med. Kerstin Breithaupt-Grögler Fachärztin für klinische Pharmakologie Frankfurt am Main e-mail: breithaupt-groegler@t-online.de -kbr-2013 16. AMG
Studieren- Erklärungen und Tipps
 Studieren- Erklärungen und Tipps Es gibt Berufe, die man nicht lernen kann, sondern für die man ein Studium machen muss. Das ist zum Beispiel so wenn man Arzt oder Lehrer werden möchte. Hat ihr Kind das
Studieren- Erklärungen und Tipps Es gibt Berufe, die man nicht lernen kann, sondern für die man ein Studium machen muss. Das ist zum Beispiel so wenn man Arzt oder Lehrer werden möchte. Hat ihr Kind das
Hilfe zur Verwendung digitaler Formulare
 Übersicht A) Allgemeines Seite 1 B) Antragstellung / Auswahl der Formulare Seite 1 Aufruf der Formulare Seite 1 Bearbeiten/Ausfüllen der Formulare Seite 2 C) Einreichen/Weiterleiten Seite 4 A) Allgemeines
Übersicht A) Allgemeines Seite 1 B) Antragstellung / Auswahl der Formulare Seite 1 Aufruf der Formulare Seite 1 Bearbeiten/Ausfüllen der Formulare Seite 2 C) Einreichen/Weiterleiten Seite 4 A) Allgemeines
proles-login. Inhalt [Dokument: L201401-1018 / v1.0 vom 16.01.2014]
![proles-login. Inhalt [Dokument: L201401-1018 / v1.0 vom 16.01.2014]](/thumbs/27/11119503.jpg "proles-login. Inhalt [Dokument: L201401-1018 / v1.0 vom 16.01.2014]") proles-login. [Dokument: L201401-1018 / v1.0 vom 16.01.2014] Inhalt 1. Einleitung 2 2. email-adresse registrieren 2 3. Benutzerinformationen des Mitarbeiters 3 4. Passwort-Rücksetzung 4 5. Passwort ändern
proles-login. [Dokument: L201401-1018 / v1.0 vom 16.01.2014] Inhalt 1. Einleitung 2 2. email-adresse registrieren 2 3. Benutzerinformationen des Mitarbeiters 3 4. Passwort-Rücksetzung 4 5. Passwort ändern
Beschreibung E-Mail Regeln z.b. Abwesenheitsmeldung und Weiterleitung
 Outlook Weiterleitungen & Abwesenheitsmeldungen Seite 1 von 6 Beschreibung E-Mail Regeln z.b. Abwesenheitsmeldung und Weiterleitung Erstellt: Quelle: 3.12.09/MM \\rsiag-s3aad\install\vnc\email Weiterleitung
Outlook Weiterleitungen & Abwesenheitsmeldungen Seite 1 von 6 Beschreibung E-Mail Regeln z.b. Abwesenheitsmeldung und Weiterleitung Erstellt: Quelle: 3.12.09/MM \\rsiag-s3aad\install\vnc\email Weiterleitung
Mitteilung der Kommission. Muster für eine Erklärung über die zur Einstufung als KMU erforderlichen Angaben (2003/C 118/03)
") 20.5.2003 Amtsblatt der Europäischen Union C 118/5 Mitteilung der Kommission Muster für eine Erklärung über die zur Einstufung als KMU erforderlichen Angaben (2003/C 118/03) Durch diese Mitteilung soll
20.5.2003 Amtsblatt der Europäischen Union C 118/5 Mitteilung der Kommission Muster für eine Erklärung über die zur Einstufung als KMU erforderlichen Angaben (2003/C 118/03) Durch diese Mitteilung soll
Teilnahme am Apple ios Developer Program
 Teilnahme am Apple ios Developer Program D-U-N-S-Nummer Für die Anmeldung als Firma brauchen Sie zunächst eine so genannte D-U-N-S-Nummer. Mehr Informationen zu dieser Nummer finden Sie unter http://de.wikipedia.org/wiki/d-u-n-s.
Teilnahme am Apple ios Developer Program D-U-N-S-Nummer Für die Anmeldung als Firma brauchen Sie zunächst eine so genannte D-U-N-S-Nummer. Mehr Informationen zu dieser Nummer finden Sie unter http://de.wikipedia.org/wiki/d-u-n-s.
Dokumentation zum Spielserver der Software Challenge
 Dokumentation zum Spielserver der Software Challenge 10.08.2011 Inhaltsverzeichnis: Programmoberfläche... 2 Ein neues Spiel erstellen... 2 Spielfeldoberfläche... 4 Spielwiederholung laden... 5 Testdurchläufe...
Dokumentation zum Spielserver der Software Challenge 10.08.2011 Inhaltsverzeichnis: Programmoberfläche... 2 Ein neues Spiel erstellen... 2 Spielfeldoberfläche... 4 Spielwiederholung laden... 5 Testdurchläufe...
Hausarzt relevante medizinische Informationen übermittelt werden, sofern der Patient damit einverstanden ist und einen Hausarzt benennt.
 Berichtspflichten von Psychologischen Psychotherapeuten und Kinder- und Jugendlichenpsychotherapeuten / Stellungnahme des Justiziars der Bundespsychotherapeutenkammer vom 25.05.04 In einem Schreiben des
Berichtspflichten von Psychologischen Psychotherapeuten und Kinder- und Jugendlichenpsychotherapeuten / Stellungnahme des Justiziars der Bundespsychotherapeutenkammer vom 25.05.04 In einem Schreiben des
zur Kreditwürdigkeitsprüfung
 EBA/GL/2015/11 19.08.2015 EBA Leitlinien zur Kreditwürdigkeitsprüfung 1 Inhaltsverzeichnis Abschnitt 1 Verpflichtung zur Einhaltung der Leitlinien und Meldepflichten 3 Abschnitt II Gegenstand, Anwendungsbereich
EBA/GL/2015/11 19.08.2015 EBA Leitlinien zur Kreditwürdigkeitsprüfung 1 Inhaltsverzeichnis Abschnitt 1 Verpflichtung zur Einhaltung der Leitlinien und Meldepflichten 3 Abschnitt II Gegenstand, Anwendungsbereich
ARCO Software - Anleitung zur Umstellung der MWSt
 ARCO Software - Anleitung zur Umstellung der MWSt Wieder einmal beschert uns die Bundesverwaltung auf Ende Jahr mit zusätzlicher Arbeit, statt mit den immer wieder versprochenen Erleichterungen für KMU.
ARCO Software - Anleitung zur Umstellung der MWSt Wieder einmal beschert uns die Bundesverwaltung auf Ende Jahr mit zusätzlicher Arbeit, statt mit den immer wieder versprochenen Erleichterungen für KMU.
PUBLIC LIMITE DE RAT DER EUROPÄISCHEN UNION. Brüssel, den 4. Mai 2007 (25.05) (OR. en) 8935/1/07 REV 1. Interinstitutionelles Dossier: 2005/0261(COD)
 (OR. en) 8935/1/07 REV 1. Interinstitutionelles Dossier: 2005/0261(COD)") Conseil UE RAT DER EUROPÄISCHEN UNION Brüssel, den 4. Mai 2007 (25.05) (OR. en) PUBLIC Interinstitutionelles Dossier: 2005/0261(COD) 8935/1/07 REV 1 LIMITE JUSTCIV 110 CODEC 421 DOKUMENT TEILWEISE ZUGÄNGLICH
Conseil UE RAT DER EUROPÄISCHEN UNION Brüssel, den 4. Mai 2007 (25.05) (OR. en) PUBLIC Interinstitutionelles Dossier: 2005/0261(COD) 8935/1/07 REV 1 LIMITE JUSTCIV 110 CODEC 421 DOKUMENT TEILWEISE ZUGÄNGLICH
Anleitung für die Lohnmeldung via ELM-Standard mittels PartnerWeb
 Ausgleichskasse Gewerbe St. Gallen Lindenstrasse 137 Postfach 245 9016 St. Gallen Telefon 071 282 29 29 Telefax 071 282 29 30 info@ahv-gewerbe.ch www.ahv-gewerbe.ch Anleitung für die Lohnmeldung via ELM-Standard
Ausgleichskasse Gewerbe St. Gallen Lindenstrasse 137 Postfach 245 9016 St. Gallen Telefon 071 282 29 29 Telefax 071 282 29 30 info@ahv-gewerbe.ch www.ahv-gewerbe.ch Anleitung für die Lohnmeldung via ELM-Standard
1 Verarbeitung personenbezogener Daten
 .WIEN WHOIS-Politik Inhalt 1 Verarbeitung personenbezogener Daten... 1 2 Zur Verwendung gesammelte Informationen... 1 3 WHOIS-Suchfunktion... 2 3.1 Einleitung... 2 3.2 Zweck... 3 3.3 Identifizieren von
.WIEN WHOIS-Politik Inhalt 1 Verarbeitung personenbezogener Daten... 1 2 Zur Verwendung gesammelte Informationen... 1 3 WHOIS-Suchfunktion... 2 3.1 Einleitung... 2 3.2 Zweck... 3 3.3 Identifizieren von
Was ist eine gute Klinische Studie - die Sicht der Statistik. Peter Martus Institut für Biometrie und Klinische Epidemiologie
 Was ist eine gute Klinische Studie - die Sicht der Statistik Peter Martus Institut für Biometrie und Klinische Epidemiologie Historisches Beispiel James Lind (1716-1794) entwickelte 1747 als britischer
Was ist eine gute Klinische Studie - die Sicht der Statistik Peter Martus Institut für Biometrie und Klinische Epidemiologie Historisches Beispiel James Lind (1716-1794) entwickelte 1747 als britischer
1. Anmeldung von Konten für das elektronische Postfach
 1. Anmeldung von Konten für das elektronische Postfach Für die Registrierung zum Elektronischen Postfach melden Sie sich bitte über die Homepage der Sparkasse Schweinfurt (www.sparkasse-sw.de) mit Ihren
1. Anmeldung von Konten für das elektronische Postfach Für die Registrierung zum Elektronischen Postfach melden Sie sich bitte über die Homepage der Sparkasse Schweinfurt (www.sparkasse-sw.de) mit Ihren
KOMMISSION DER EUROPÄISCHEN GEMEINSCHAFTEN. Vorschlag für GEMEINSAME REGELUNG
 KOMMISSION DER EUROPÄISCHEN GEMEINSCHAFTEN Brüssel, den 15.4.2004 SEK(2004) 411 endgültig Vorschlag für GEMEINSAME REGELUNG zur Festlegung der Modalitäten für die Überweisung eines Teils der Dienstbezüge
KOMMISSION DER EUROPÄISCHEN GEMEINSCHAFTEN Brüssel, den 15.4.2004 SEK(2004) 411 endgültig Vorschlag für GEMEINSAME REGELUNG zur Festlegung der Modalitäten für die Überweisung eines Teils der Dienstbezüge
Der Schutz von Patientendaten
 Der Schutz von Patientendaten bei (vernetzten) Software-Medizinprodukten aus Herstellersicht 18.09.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
Der Schutz von Patientendaten bei (vernetzten) Software-Medizinprodukten aus Herstellersicht 18.09.2014 Gerald Spyra, LL.M. Kanzlei Spyra Vorstellung meiner Person Gerald Spyra, LL.M. Rechtsanwalt Spezialisiert
SharePoint Demonstration
 SharePoint Demonstration Was zeigt die Demonstration? Diese Demonstration soll den modernen Zugriff auf Daten und Informationen veranschaulichen und zeigen welche Vorteile sich dadurch in der Zusammenarbeit
SharePoint Demonstration Was zeigt die Demonstration? Diese Demonstration soll den modernen Zugriff auf Daten und Informationen veranschaulichen und zeigen welche Vorteile sich dadurch in der Zusammenarbeit
Vereinbarung über privatzahnärztliche Leistungen bei der kieferorthopädischen Behandlung
 Vereinbarung über privatzahnärztliche Leistungen bei der kieferorthopädischen Behandlung Zwischen Zahlungspflichtige/-r und Zahnärztin I Zahnarzt für Patient (falls abweichend vom Zahlungspflichtigen)
Vereinbarung über privatzahnärztliche Leistungen bei der kieferorthopädischen Behandlung Zwischen Zahlungspflichtige/-r und Zahnärztin I Zahnarzt für Patient (falls abweichend vom Zahlungspflichtigen)
Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt?
 DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
DGSV-Kongress 2009 Wann ist eine Software in Medizinprodukte- Aufbereitungsabteilungen ein Medizinprodukt? Sybille Andrée Betriebswirtin für und Sozialmanagement (FH-SRH) Prokuristin HSD Händschke Software
Informationen zum Thema Europäische Krankenversicherungskarte
 Gesundheitskarte AKTUELL Informationen zum Thema Europäische Krankenversicherungskarte Von Anfang an ist die Rückseite der elektronischen Gesundheitskarte für die Aufnahme der Europäischen Krankenversicherungskarte
Gesundheitskarte AKTUELL Informationen zum Thema Europäische Krankenversicherungskarte Von Anfang an ist die Rückseite der elektronischen Gesundheitskarte für die Aufnahme der Europäischen Krankenversicherungskarte
Einleitende Bemerkungen
 Einleitende Bemerkungen EU-FORMBLATT LENKFREIE TAGE / KONTROLLGERÄT MANUELLER NACHTRAG ENTSCHEIDUNGSHILFE FÜR FAHRPERSONAL VON VERORDNUNGS-FAHRZEUGEN 1 BEI TÄTIGKEITEN IM INNERSTAATLICHEN VERKEHR Zur Frage,
Einleitende Bemerkungen EU-FORMBLATT LENKFREIE TAGE / KONTROLLGERÄT MANUELLER NACHTRAG ENTSCHEIDUNGSHILFE FÜR FAHRPERSONAL VON VERORDNUNGS-FAHRZEUGEN 1 BEI TÄTIGKEITEN IM INNERSTAATLICHEN VERKEHR Zur Frage,
Nutzung dieser Internetseite
 Nutzung dieser Internetseite Wenn Sie unseren Internetauftritt besuchen, dann erheben wir nur statistische Daten über unsere Besucher. In einer statistischen Zusammenfassung erfahren wir lediglich, welcher
Nutzung dieser Internetseite Wenn Sie unseren Internetauftritt besuchen, dann erheben wir nur statistische Daten über unsere Besucher. In einer statistischen Zusammenfassung erfahren wir lediglich, welcher
Methodische Fragen zur frühen Nutzenbewertung nach 35a SGB V
 Die frühe Nutzenbewertung nach AMNOG Rechtssymposium des G-BA, Berlin, 16. Nov. 2010 Methodische Fragen zur frühen Nutzenbewertung nach 35a SGB V Jürgen Windeler AMNOG Nutzenbewertung für Arzneimittel
Die frühe Nutzenbewertung nach AMNOG Rechtssymposium des G-BA, Berlin, 16. Nov. 2010 Methodische Fragen zur frühen Nutzenbewertung nach 35a SGB V Jürgen Windeler AMNOG Nutzenbewertung für Arzneimittel
Betroffenes Produkt: PRI Femureinschlägerkopf, 00-5901-032-00 (vollständige Liste der betroffenen Chargen siehe Anhang 1)
") 22. Mai 2015 An: Betrifft: Risikomanager und Chirurgen DRINGENDER CHARGENSPEZIFISCHER RÜCKRUF EINES MEDIZINPRODUKTES Betroffenes Produkt: PRI Femureinschlägerkopf, 00-5901-032-00 (vollständige Liste der
22. Mai 2015 An: Betrifft: Risikomanager und Chirurgen DRINGENDER CHARGENSPEZIFISCHER RÜCKRUF EINES MEDIZINPRODUKTES Betroffenes Produkt: PRI Femureinschlägerkopf, 00-5901-032-00 (vollständige Liste der
Änderungen ISO 27001: 2013
 Änderungen ISO 27001: 2013 Loomans & Matz AG August-Horch-Str. 6a, 55129 Mainz Deutschland Tel. +496131-3277 877; www.loomans-matz.de, info@loomans-matz.de Die neue Version ist seit Oktober 2013 verfügbar
Änderungen ISO 27001: 2013 Loomans & Matz AG August-Horch-Str. 6a, 55129 Mainz Deutschland Tel. +496131-3277 877; www.loomans-matz.de, info@loomans-matz.de Die neue Version ist seit Oktober 2013 verfügbar
Fragen und Antworten zur UV-Jahresmeldung nach 28a Abs. 2a SGB IV
 Fragen und Antworten zur UV-Jahresmeldung nach 28a Abs. 2a SGB IV Mit dem Fünften Gesetz zur Änderung des Vierten Buches Sozialgesetzbuch und anderer Gesetze (5. SGB IV-ÄndG) vom 15.04.2015 (BGBl. 2015
Fragen und Antworten zur UV-Jahresmeldung nach 28a Abs. 2a SGB IV Mit dem Fünften Gesetz zur Änderung des Vierten Buches Sozialgesetzbuch und anderer Gesetze (5. SGB IV-ÄndG) vom 15.04.2015 (BGBl. 2015
BüroWARE Exchange Synchronisation Grundlagen und Voraussetzungen
 BüroWARE Exchange Synchronisation Grundlagen und Voraussetzungen Stand: 13.12.2010 Die BüroWARE SoftENGINE ist ab Version 5.42.000-060 in der Lage mit einem Microsoft Exchange Server ab Version 2007 SP1
BüroWARE Exchange Synchronisation Grundlagen und Voraussetzungen Stand: 13.12.2010 Die BüroWARE SoftENGINE ist ab Version 5.42.000-060 in der Lage mit einem Microsoft Exchange Server ab Version 2007 SP1
- 1- Bundesministerium für Gesundheit und Soziale Sicherung
 - 1- Bundesanzeiger vom 25. März 2004, S. 6104 Bundesministerium für Gesundheit und Soziale Sicherung Bekanntmachung von Empfehlungen zum Versandhandel und elektronischen Handel mit Arzneimitteln Vom 18.
- 1- Bundesanzeiger vom 25. März 2004, S. 6104 Bundesministerium für Gesundheit und Soziale Sicherung Bekanntmachung von Empfehlungen zum Versandhandel und elektronischen Handel mit Arzneimitteln Vom 18.
Praktischer Leitfaden für eine angemessene Versorgung
 Mein Recht als Patient Praktischer Leitfaden für eine angemessene Versorgung Gesundheit ist ein Menschenrecht Im Grundgesetz ist das Recht auf körperliche Unversehrtheit fest verankert. Damit hat unser
Mein Recht als Patient Praktischer Leitfaden für eine angemessene Versorgung Gesundheit ist ein Menschenrecht Im Grundgesetz ist das Recht auf körperliche Unversehrtheit fest verankert. Damit hat unser
E-Government Sondertransporte (SOTRA) Registrierung von Benutzerkennung
 Registrierung von Benutzerkennung") E-Government Sondertransporte (SOTRA) Registrierung von Benutzerkennung Projektteam Sondertransporte Land OÖ Version September 2012 Alle Rechte, insbesondere das Recht der Vervielfältigung, Verbreitung
E-Government Sondertransporte (SOTRA) Registrierung von Benutzerkennung Projektteam Sondertransporte Land OÖ Version September 2012 Alle Rechte, insbesondere das Recht der Vervielfältigung, Verbreitung
Fragen und Antworten zur Prüfmöglichkeit für ausländische Investitionen (Änderung des Außenwirtschaftsgesetzes und der Außenwirtschaftsverordnung)
") Fragen und Antworten zur Prüfmöglichkeit für ausländische Investitionen (Änderung des Außenwirtschaftsgesetzes und der Außenwirtschaftsverordnung) 1. Welche Investitionen können geprüft werden? Einer Prüfung
Fragen und Antworten zur Prüfmöglichkeit für ausländische Investitionen (Änderung des Außenwirtschaftsgesetzes und der Außenwirtschaftsverordnung) 1. Welche Investitionen können geprüft werden? Einer Prüfung
Sie haben das Recht, binnen vierzehn Tagen ohne Angabe von Gründen diesen Vertrag zu widerrufen.
 Widerrufsbelehrung Nutzt der Kunde die Leistungen als Verbraucher und hat seinen Auftrag unter Nutzung von sog. Fernkommunikationsmitteln (z. B. Telefon, Telefax, E-Mail, Online-Web-Formular) übermittelt,
Widerrufsbelehrung Nutzt der Kunde die Leistungen als Verbraucher und hat seinen Auftrag unter Nutzung von sog. Fernkommunikationsmitteln (z. B. Telefon, Telefax, E-Mail, Online-Web-Formular) übermittelt,