Das Szenario...ein neues tödliches Virus!
|
|
|
- Hildegard Winkler
- vor 6 Jahren
- Abrufe
Transkript

1
2 Das Szenario...ein neues tödliches Virus! Labor: Isolierung der Erbsubstanz und Sequenzierung Computer: Erkennen der Virusgene (de novo Genvorhersage) Ähnlichkeit zu bekannten Genen? (Datenbanksuchen) Verwandtschaft? Ausbreitung? Herkunft? (Phylogenetische Rekonstruktion) Struktur der Proteine? (Struktur-Vorhersage, Modellierung) Wirkstoff-Design Labor: Wirkstoff-Test
 Ausbreitung von Lebewesen (Anthropologie, Ökologie,")
3 Molekulare Phylogenie Verwandtschaft von Organismen (molekulare Systematik, Forensik) Verwandschaft zwischen Genen/Proteinen (Genomevolution, Gen/Proteinfunktion) Ausbreitung von Lebewesen (Anthropologie, Ökologie, Epidemiologie)
 (5")
 Das")






4 Grundlage! (1.500 MYA) (5 MYA) (100 MYA) Das Leben ist nur einmal enstanden. => alle Organismen sind miteinander verwandt, d.h. haben einen Vorfahren, der in der Vergangenheit gelebt hat. Dan Graur 4 Dan Graur 4

")

5 Grundbegriffe Innengruppe (ingroup) A B C D E Ast (branch) Dichotomie Polytomie A B C D E Wurzel (root) Knotenpunkt (node) 5
6 Stammbaum-Typen Ohne Außengruppe: Mit Außengruppe: Neunauge Hai Hai Maus Neunauge Flösselhecht Goldfisch Zebrafisch Strahlenflosser Mensch Flösselhecht Forelle Lungenfisch Molch Forelle Molch Ochsenfrosch Ochsen- frosch Krallenfrosch Zebrafisch Goldfisch Krallenfrosch Maus Landwirbeltiere Lungenfisch Mensch Evolutionsrichtung? Evolutionsrichtung 6

 weitgehend frei von Interpretationseinflüssen ( reduziert, etwas abgeflacht etc.")
7 Molekulare vs. klassische Phylogenie Sequenzen sind direkt vererbt; keine Umwelteinflüsse Sequenzdaten sind in großer Menge relativ kostengünstig und schnell zu erhalten (Dank sei der PCR!!!) weitgehend frei von Interpretationseinflüssen ( reduziert, etwas abgeflacht etc.) Sequenzen erlauben Vergleiche über große Distanzen (Tiere, Pilze, Pflanzen) Dennoch: auch molekulare Daten können zu falschen Stammbäumen führen!


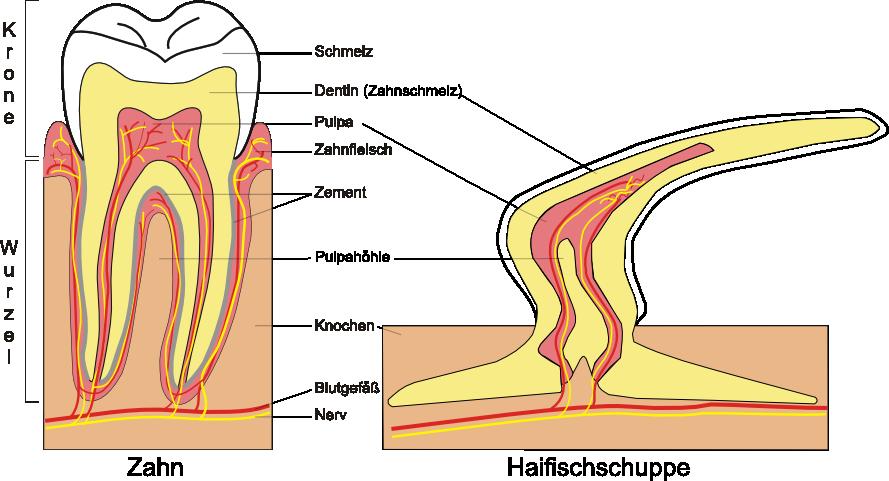
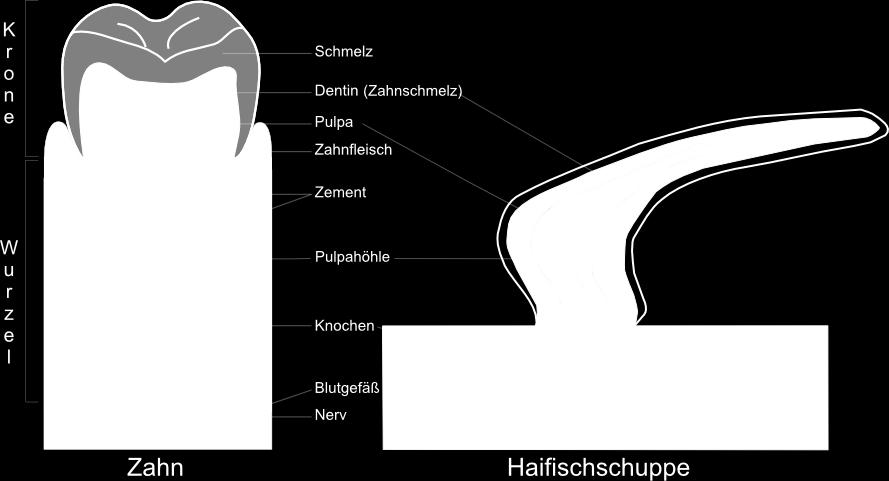


8 Welche Vergleiche mache Sinn?

9 Welche Vergleiche mache Sinn? HOMOLOGIE heißt das Zauberwort für sinnvolle Vergleiche!
10 Welche Vergleiche mache Sinn? PlePPO YWREDFGINSHHWHWHLVYPIEM-----NVNRDRKGELFYYMHQQMVARYDWERLSVNLNRVEKLE PlePPO PmoPPO YWREDYGINVHHWHWHLIYPPAM-----GFDRDRKGELFYYMHQQVIARYDIERLCLGLPKVEKLD PmoPPO BmoPPO1 YFREDIGINLHHWHWHLVYPFDAADRA-IVNKDRRGELFYYMHQQIIARYNVERMCNNLSRVRRYN BmoPPO DmePPOA1 YFREDIGVNSHHWHWHLVYPTTGPTE--VVNKDRRGELFYYMHHQILARYNVERFCNNLKKVQPLN DmePPOA DmePPO2 YFREDLGINLHHWHWHLVYPFEASDRS-IVAKDRRGELFYYMHQQVIARYNAERFSNNLARVLPFN DmePPO DmePPO3 YFREDLGVNLHHWHWHLVYPIEAPDRS-IVDKDRRGELFYYMHQQIIARYNAERLSNHMARVQPFN DmePPO3 65? 65 EcaHcA YFREDIGVNAHHWHWHVVYPSTYDPAFFGKVKDRKGELFYYMHQQMCARYDCERLSNGLNRMIPFH EcaHcA EcaHcD YFREDIGINSHHWHWHLVYPAFYDADIFGKIKDRKGELFYYMHQQMCARYDCERLSVGLQRMIPFQ EcaHcD EcaHcF YFREDIGANAHHWHWHIVYPPTWDASVMSKVKDRKGELFYYMHQQMCARYDCDRLSTGLRRMIPFH EcaHcF LpoHc2 YYREDVGINAHHWHWHLVYPSTWNPKYFGKKKDRKGELFYYMHQQMCARYDCERLSNGMHRMLPFN LpoHc PvaHc YFGEDIGLNTHHVTWHMEFPFWWNDAYG-HHLDRKGENFFWIHHQLTVRFDAERLSNYLDPVGELQ PvaHc PirHcC YFGEDVGMNTHHVLWHMEFPFWWEDSSG-RHLDRKGESFFWVHHQLTVRYDAERLSNHLDPVEELS PirHcC PirHcA YFGEDIGMNIHHVTWHMDFPFWWEDSYG-YHLDRKGELFFWVHHQLTARFDFERLSNWLDPVDELH PirHcA PlePPO YWREDFGINSHHWHWHLVYPIEM-----NVNRDRKGELFYYMHQQMVARYDWERLSVNLNRVEKLE 61 PmoPPO YWREDYGINVHHWHWHLIYPPAM-----GFDRDRKGELFYYMHQQVIARYDIERLCLGLPKVEKLD 61 BmoPPO1 YFREDIGINLHHWHWHLVYPFDAADRA-IVNKDRRGELFYYMHQQIIARYNVERMCNNLSRVRRYN 65 DmePPOA1 YFREDIGVNSHHWHWHLVYPTTGPTE--VVNKDRRGELFYYMHHQILARYNVERFCNNLKKVQPLN 64 DmePPO2 YFREDLGINLHHWHWHLVYPFEASDRS-IVAKDRRGELFYYMHQQVIARYNAERFSNNLARVLPFN 65 DmePPO3 YFREDLGVNLHHWHWHLVYPIEAPDRS-IVDKDRRGELFYYMHQQIIARYNAERLSNHMARVQPFN 65 EcaHcA YFREDIGVNAHHWHWHVVYPSTYDPAFFGKVKDRKGELFYYMHQQMCARYDCERLSNGLNRMIPFH 66 EcaHcD YFREDIGINSHHWHWHLVYPAFYDADIFGKIKDRKGELFYYMHQQMCARYDCERLSVGLQRMIPFQ 66 EcaHcF YFREDIGANAHHWHWHIVYPPTWDASVMSKVKDRKGELFYYMHQQMCARYDCDRLSTGLRRMIPFH 66 LpoHc2 YYREDVGINAHHWHWHLVYPSTWNPKYFGKKKDRKGELFYYMHQQMCARYDCERLSNGMHRMLPFN 66 PvaHc YFGEDIGLNTHHVTWHMEFPFWWNDAYG-HHLDRKGENFFWIHHQLTVRFDAERLSNYLDPVGELQ 65 PirHcC YFGEDVGMNTHHVLWHMEFPFWWEDSSG-RHLDRKGESFFWVHHQLTVRYDAERLSNHLDPVEELS 65 PirHcA YFGEDIGMNIHHVTWHMDFPFWWEDSYG-YHLDRKGELFFWVHHQLTARFDFERLSNWLDPVDELH 65
11 Allgemeine Vorgehensweise Sequenz 1 Sequenz 1: KIADKNFTYRHHNQLV Sequenz 4 Sequenz 2: KVAEKNMTFRRFNDII Sequenz 2 Sequenz 3: KIADKDFTYRHW-QLV Sequenz 4: KVADKNFSYRHHNNVV Sequenz 3 Sequenz 5: KLADKQFTFRHH-QLV Sequenz 5 Multiples Sequenzalignment erstellen (DNA oder Protein) Sequenzen vergleichen > Ähnlichkeit bestimmen Aus Ähnlichkeitsmaß die Verwandtschaft rekonstruieren (Baum)
12 Phylogenie-Rekonstruktion ist kein triviales Problem mit 3 Taxa kann man 1 unverwurzelten Baum erstellen, aber 3 alternative Bäume mit Wurzel es ist viel leichter und sicherer, einen unrooted Baum zu erstellen: d. h. nur dann rooten, wenn die Outgroup klar definiert ist
13 Phylogenie-Rekonstruktion ist kein triviales Problem



14 Nochmal die Frage... Wann DNA? Wann Protein? Eng verwandte SARS-Varianten in der menschlichen Population Corona-Virus-Gruppen aus verschiedenen Spezies
15 Ein Alignment ist immer eine Hypothese zur Sequenzevolution! NAYLS NAKYLS NAFS NAFLS SeqA N A F L S SeqB N A F - S SeqC N A K Y L S SeqD N A Y L S +K -L Y -> F Wir waren ja leider nicht direkt dabei...!
16 Warum ist es nicht einfach, das beste Alignment zu konstruieren? 2 Sequenzen à 300 Bp = mögliche Alignments!!! Computer-Algorithmen erforderlich, die ohne Ausprobieren aller Möglichkeiten auskommen. Regelwerk notwendig, um bestmögliches Alignment zu erkennen
17 Warum ist es nicht einfach, das beste Alignment zu konstruieren? seqa TCAGACGATTG (11) seqb TCGGAGCTG (9) I. TCAG-ACG-ATTG TC-GGA-GC-T-G Annahmen über den Ablauf der Sequenz-Evolution: I. Keine mismatches II. III. TCAGACGATTG TCGGAGCTG-- TCAG-ACGATTG TC-GGA--GCTG II. Keine internen Lücken III. Von beidem Etwas Aber was ist richtig?

18 Überlegungen... Jede Sequenz lässt sich mit einer jeden anderen Sequenz alignen! Aber macht das Alignment auch Sinn? Also: Haben wir die richtigen Annahmen über den Verlauf der Evolution getroffen?? Wir brauchen evolutionäre Modelle, um ein möglichst richtiges Alignment zu erstellen!
19 Was bedeutet es, ein Evolutionsmodell zu haben? Bov Co-V SARS Mur HepV Ein Evolutionsmodell basiert auf empirischen Daten! Zum Beispiel: Ich weiß, die Aminosäure Cystein ist für die Proteinstruktur äußerst wichtig! " Cysteine sind also konserviert während der Evolution von Proteinen! " Cysteine können daher beim Alignment zweier Proteinsequenzen als Ankerpunkte dienen " ein Alignment mit übereinanderstehenden Cysteinen würde danach mit Pluspunkten belohnt

20 SARS: konservierte Cysteine im Alignment des spike-proteins Eickmann et al Resultat: Verwandtschaft von SARS zu Gruppe 2-Coronaviren?

21 Ein einfacher Score-Wert zur Bewertung eines Alignments S = Y - W k x Z k S = Similarity-Score ( Belohnungspunkte ) Y = Anzahl an Matches Zk = Anzahl der gaps mit Länge k Wk = gap penalty für gaps der Länge k Mit Setzen der gap penalty trifft man Annahmen über die relative Häufigkeit von indel-mutationen während der Evolution!

22 Eine einfache Substitutionsmatrix für Nukleotidsequenzen A C G T A C G T alle Richtungen von Nt-Austauschen sind gleich wahrscheinlich bei jedem match beider Sequenzen gibt es 1 Belohnungspunkt für den Übereinstimmungs-Score
 unterschiedlich hoch! Definition der Kosten/Belohnungen erfolgt über Matrizen: z. B. PAM-Matrizen (Dayhoff 1978)")
23 Substitutions-Matrizen für Proteine bei Proteinen gibt es 20 As! Arg Lys chemisch-funktionelle Ähnlichkeit bestimmt Wahrscheinlichkeit eines Austauschs während der Evolution. Daher......sind die Kosten für bestimmte Austausche (bzw. die Belohnung für gleiche As) unterschiedlich hoch! Definition der Kosten/Belohnungen erfolgt über Matrizen: z. B. PAM-Matrizen (Dayhoff 1978)

24 PAM-Matrizen... Margaret Dayhoff...definieren Belohnungswerte für zwei Aminosäuren, die sich in einem Alignment gegenüberstehen: positiver Wert = Aminosäuren, die sich häufig in Alignments gegenüberstehen und somit funktionell konserviert sind z.b. W-W C -C aber W-V -6


25 Wir haben also Kriterien (Substitutionsmatrizen, gap penalties), um Alignments zu bewerten. Aber wie werden Alignments überhaupt erstellt?

26 Needleman-Wunsch (N-W) 1970 Bei Erstellung des Alignments werden zunächst kleine Problem-Schritte gelöst. Dann wird aus den Teillösungen das Gesamtalignment rekonstruiert... Algorithmus: dynamic programming
27 Needleman-Wunsch
28 Needleman-Wunsch es wird zunächst eine zweidimensionale Matrix mit den beiden zu vergleichenden Sequenzen erstellt in die Zellen der Matrix wird der Alignment-Score für die jeweils verglichenen Sequenzpositionen hineingeschrieben. Die Berechnung des Score-Werts erfolgt natürlich anhand einer Substitutionsmatrize. das Alignment ergibt sich als Pfad durch die Matrix. Der Pfad mit der höchsten Endsumme gewinnt...
29 N-W ist eine exakte Vorgehensweise und viel zu aufwändig für multiple Sequenzvergleiche! Wir brauchen... Heuristik (altgr. εὑρίσκω heurísko ich finde ; von εὑρίσκειν heurískein auffinden, entdecken ) bezeichnet die Kunst, mit begrenztem Wissen (unvollständigen Informationen) und wenig Zeit dennoch zu wahrscheinlichen Aussagen oder praktikablen Lösungen zu kommen. [1] Es bezeichnet ein analytisches Vorgehen, bei dem mit begrenztem Wissen über ein System mit Hilfe mutmaßender Schlussfolgerungen Aussagen über das System getroffen werden. Die damit gefolgerten Aussagen können von der optimalen Lösung abweichen.
30 Progressives Alignment 1) Sequenzvergleich A B C D Alle Sequenzen werden miteinander verglichen (Option A: schnelles "quick and dirty" Alignment Option B: exaktes, langsames Needleman-Wunsch) => Berechnen der Distanzen 30
31 Progressives Alignment 2) Ähnliche Sequenzen werden gruppiert => Cluster-Analyse = Erstellung eines hierarchischen Stammbaums ("guide tree"). A B A - B C D A D B C C D "guide tree" 31
32 Progressives Alignment 3) Alignment von nahe verwandten Sequenzen; die ähnlichsten zuerst. A D B C A D B C 32
33 Progressives Alignment 4) Sukzessives globales Alignment A D A D B C alte gaps erhalten, neue hinzugefügt B C A D B C 33
34 Progressives Alignment 1. paarweiser Vergleich aller Sequenzen miteinander => Berechnung der Distanzen zweier Sequenzen 2. gruppiert Sequenzen nach Ähnlichkeit (Cluster- Bildung) 3. Erstellung paarweiser Alignments 4. sukzessives Alignment nach Ähnlichkeit, dabei die ähnlichsten Sequenzpaare zuerst Feng & Doolittle (1987): PileUp (GCG package) Higgins and Sharp (1988), Thompson et al. (1994): CLUSTAL Notredame et al. (2000): T-COFFEE 34
35 Zur Erinnerung: Allgemeine Vorgehensweise Sequenz 1 Sequenz 1: KIADKNFTYRHHNQLV Sequenz 4 Sequenz 2: KVAEKNMTFRRFNDII Sequenz 2 Sequenz 3: KIADKDFTYRHW-QLV Sequenz 4: KVADKNFSYRHHNNVV Sequenz 3 Sequenz 5: KLADKQFTFRHH-QLV Sequenz 5 Multiples Sequenzalignment erstellen (DNA oder Protein) Sequenzen vergleichen > Ähnlichkeit bestimmen Aus Ähnlichkeitsmaß die Verwandtschaft rekonstruieren (Baum)
36 Vom Alignment zu einem einfachen Baum-Rekonstruktionsverfahren Aus dem Alignment ergibt sich zunächst, wie ähnlich oder unähnlich die Sequenzen zueinander sind. Meist wird eine Distanzmatrix erstellt: A B C D OTU * A OTU B OTU C 0 19 OTU D 0 * OTU = operational taxonomic unit: z. B. Spezies, Gen, Protein
 der")
37 Vom Alignment zu einem einfachen Baum-Rekonstruktionsverfahren Sokal and Michener 1967! UPGMA = Unweighted Pair-Group Method using Arithmetric Means eine Methode, die auf der Berechnung von Unähnlichkeiten (Distanzen) der alignierten Sequenzen beruht ( Distanz-Methode )
38 UPGMA 1. Ausgangs-Distanz-Matrix A B C D OTU A OTU B OTU C 0 19 OTU D A B 2. Neu berechnete Distanz-Matrix A/B C D OTU A/B OTU C 0 19 OTU D 0 (AC + BC) / A B C
39 UPGMA 3. Neu berechnete Distanz-Matrix A/B/C D Sequenz A/B/C 0 19 Sequenz D A B 5.5 C 9.5 D
40 UPGMA
41 UPGMA-Problem Ausgangsmatrix A B C D OTU A OTU B OTU C 0 19 OTU D 0 rekonstruierte Matrix A B C D OTU A OTU B OTU C 0 19 OTU D A B 5.5 C D Astlängen des UPGMA-Baums passen nicht exakt zur Realität...

42 Practical Exercises 4 Build a phylogenetic tree manually using UPGMA Produce multiple nucleotide and amino acid sequence alignment of SARS genes and proteins Where did SARS originate and how did it spread in the population? > Prepare a nt-based tree from different SARS genomes Which are the distant relatives of SARS coronavirus? > Prepare an aa-based tree for the spike protein

43 SARS-Verwandschaft RNA Polymerase (As) Spike Protein (As) Unterschiedliche Datensätze und Rekonstruktionsmethoden können leicht unterschiedliche Baum-Topologien ergeben!! Aber: SARS Co-V ist alter Verwandter der Gruppe 2 Coronaviren

44 SARS- Phylogenie: Ausbreitung
45 SARS-Phylogenie: Herkunft? DNA (Komplettgenom) Varianten sind >99% identisch. Dennoch ist eine geographische Zuordnung möglich. Sequenz zeigt Besonderheit: Sein Spike-Gen hat Insertion von 29 Bp, die sonst nur in tierischen SARS-Verwandten gefunden worden ist!
46 SARS-Phylogenie: Herkunft?
 Marderhund - Raccoon dog (Nyctereutes")
47 SARS: zoonotischer Ursprung Larvenroller - palm civet (Paguma larvata) Marderhund - Raccoon dog (Nyctereutes procyonoides)
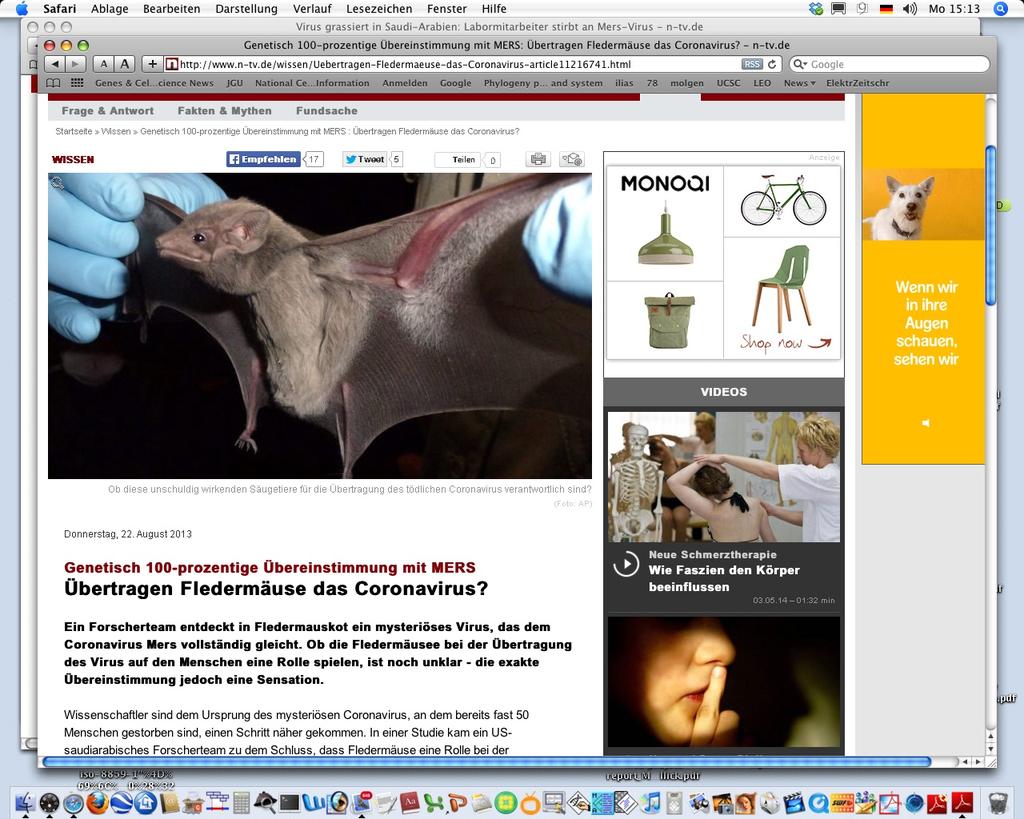
48
 Labor: Wirkstoff-Test Ähnlichkeit zu bekannten Genen?")
49 Das Szenario...ein neues tödliches Virus! Labor: Isolierung der Erbsubstanz und Sequenzierung Computer: Erkennen der Virusgene (de novo Genvorhersage) Labor: Wirkstoff-Test Ähnlichkeit zu bekannten Genen? (Datenbanksuchen) Verwandtschaft? Ausbreitung? Herkunft? (Phylogenetische Rekonstruktion) Struktur der Proteine? (Struktur-Vorhersage, -Modellierung) Wirkstoff-Design
Alignment von DNA- und Proteinsequenzen
 WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld. Institut für Zoologie Universität Mainz
 Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) Daten einen Stammbaum, und was sagt mir
Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) Daten einen Stammbaum, und was sagt mir
Sequenz Alignment Teil 2
 Sequenz Alignment Teil 2 14.11.03 Vorlesung Bioinformatik 1 Molekulare Biotechnologie Dr. Rainer König Besonderen Dank an Mark van der Linden, Mechthilde Falkenhahn und der Husar Biocomputing Service Gruppe
Sequenz Alignment Teil 2 14.11.03 Vorlesung Bioinformatik 1 Molekulare Biotechnologie Dr. Rainer König Besonderen Dank an Mark van der Linden, Mechthilde Falkenhahn und der Husar Biocomputing Service Gruppe
Multiples Sequenzalignment
 WS2013/2014 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln HsaHBA GAEALERMFLSFPTTKTYF HsaHBZ GTETLERLFLSHPQTKTYF HsaHBE GGEALGRLLVVYPWTQRFF HsaHBG GGETLGRLLVVYPWTQRFF
WS2013/2014 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln HsaHBA GAEALERMFLSFPTTKTYF HsaHBZ GTETLERLFLSHPQTKTYF HsaHBE GGEALGRLLVVYPWTQRFF HsaHBG GGETLGRLLVVYPWTQRFF
Fernstudium "Molekulare Evolution" Bernhard Lieb Michael Schaffeld. Institut für Zoologie Universität Mainz
 Fernstudium "Molekulare Evolution" ernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) aten einen Stammbaum, und was sagt mir
Fernstudium "Molekulare Evolution" ernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) aten einen Stammbaum, und was sagt mir
MOL.504 Analyse von DNA- und Proteinsequenzen. Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
Bioinformatik. Lokale Alignierung Gapkosten. Silke Trißl / Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
Bioinformatik Lokale Alignierung Gapkosten Silke Trißl / Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Ähnlichkeit Lokales und globales Alignment Gapped Alignment Silke Trißl:
Homologie und Sequenzähnlichkeit. Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Studiengang Informatik der FH Gießen-Friedberg. Sequenz-Alignment. Jan Schäfer. WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel
 Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
Studiengang Informatik der FH Gießen-Friedberg Sequenz-Alignment Jan Schäfer WS 2006/07 Betreuer: Prof. Dr. Klaus Quibeldey-Cirkel Überblick Einführung Grundlagen Wann ist das Merkmal der Ähnlichkeit erfüllt?
Threading - Algorithmen
 Threading - Algorithmen Florian Lindemann 22.11.2007 Florian Lindemann () Threading - Algorithmen 22.11.2007 1 / 25 Gliederung 1 Prospect Scoring Function Algorithmus Weitere Eigenschaften Komplexität
Threading - Algorithmen Florian Lindemann 22.11.2007 Florian Lindemann () Threading - Algorithmen 22.11.2007 1 / 25 Gliederung 1 Prospect Scoring Function Algorithmus Weitere Eigenschaften Komplexität
Bioinformatik. Substitutionsmatrizen BLAST. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Bioinformatik Substitutionsmatrizen BLAST Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Substitutionsmatrizen: PAM und BLOSSUM Suche in Datenbanken: Basic Local Alignment Search
Molekulare Phylogenie
 Molekulare Phylogenie Grundbegriffe Methoden der Stammbaum-Rekonstruktion Thomas Hankeln, Institut für Molekulargenetik SS 2010 Grundlagen der molekularen Phylogenie Evolution äußert sich durch Veränderungen
Molekulare Phylogenie Grundbegriffe Methoden der Stammbaum-Rekonstruktion Thomas Hankeln, Institut für Molekulargenetik SS 2010 Grundlagen der molekularen Phylogenie Evolution äußert sich durch Veränderungen
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
Darwins Erben - Phylogenie und Bäume
 Vorlesung Einführung in die Bioinforma4k SoSe2011 Darwins Erben - Phylogenie und Bäume Prof. Daniel Huson ZBIT Center for Bioinformatics Center for Bioinformatics Charles Darwin und Bäume Darwin's Notizbuch
Vorlesung Einführung in die Bioinforma4k SoSe2011 Darwins Erben - Phylogenie und Bäume Prof. Daniel Huson ZBIT Center for Bioinformatics Center for Bioinformatics Charles Darwin und Bäume Darwin's Notizbuch
Vorlesung Einführung in die Bioinformatik
 Vorlesung Einführung in die Bioinformatik Dr. Stephan Weise 04.04.2016 Einführung Gegeben: zwei Sequenzen Gesucht: Ähnlichkeit quantitativ erfassen Entsprechungen zwischen einzelnen Bausteinen beider Sequenzen
Vorlesung Einführung in die Bioinformatik Dr. Stephan Weise 04.04.2016 Einführung Gegeben: zwei Sequenzen Gesucht: Ähnlichkeit quantitativ erfassen Entsprechungen zwischen einzelnen Bausteinen beider Sequenzen
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de") Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
V3 - Multiples Sequenz Alignment und Phylogenie
 V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 3. Vorlesung SS 2011 Softwarewerkzeuge der Bioinformatik
V3 - Multiples Sequenz Alignment und Phylogenie Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk 3. Vorlesung SS 2011 Softwarewerkzeuge der Bioinformatik
Lokale Sequenzähnlichkeit. Genomische Datenanalyse 9. Kapitel
 Lokale Sequenzähnlichkeit Genomische Datenanalyse 9. Kapitel Globale Sequenzähnlichkeit: Zwei Cytochrome C Sequenzen: Eine vom Menschen und eine aus der Maus. Die Sequenzen sind gleich lang, man kann sie
Lokale Sequenzähnlichkeit Genomische Datenanalyse 9. Kapitel Globale Sequenzähnlichkeit: Zwei Cytochrome C Sequenzen: Eine vom Menschen und eine aus der Maus. Die Sequenzen sind gleich lang, man kann sie
Phylogenetische Analyse
 Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Phylogenetik. Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann. Webseite zur Vorlesung
 Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
Phylogenetische Rekonstrukion Sparsamkeits- und Abstandsmethoden 5. Juli 2012
 Merle Erpenbeck Phylogenetische Rekonstrukion Sparsamkeits- und Abstandsmethoden 5. Juli 202 Seminarausarbeitung im Seminar Mathematische Biologie vorgelegt von Merle Erpenbeck Matrikelnummer: 5896 Betreuer:
Merle Erpenbeck Phylogenetische Rekonstrukion Sparsamkeits- und Abstandsmethoden 5. Juli 202 Seminarausarbeitung im Seminar Mathematische Biologie vorgelegt von Merle Erpenbeck Matrikelnummer: 5896 Betreuer:
Algorithmische Bioinformatik 1
 Algorithmische Bioinformatik 1 Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2009 Übersicht Paarweises
Algorithmische Bioinformatik 1 Dr. Hanjo Täubig Lehrstuhl für Effiziente Algorithmen (Prof. Dr. Ernst W. Mayr) Institut für Informatik Technische Universität München Sommersemester 2009 Übersicht Paarweises
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse
 Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
MOL.504 Analyse von DNA- und Proteinsequenzen
 MOL.504 Analyse von DNA- und Proteinsequenzen Kurs 1 Monika Oberer, Karl Gruber MOL.504 Modul-Übersicht Einführung, Datenbanken BLAST-Suche, Sequenzalignment Proteinstrukturen Virtuelles Klonieren Abschlusstest
MOL.504 Analyse von DNA- und Proteinsequenzen Kurs 1 Monika Oberer, Karl Gruber MOL.504 Modul-Übersicht Einführung, Datenbanken BLAST-Suche, Sequenzalignment Proteinstrukturen Virtuelles Klonieren Abschlusstest
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag
 WS2013/2014 Woche 3 - Montag") VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
BLAST. Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02
 BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
Einführung in die Bioinformatik
 Enno Abteilung Theoretische Informatik Universität Ulm October 18, 2016 Überblick 1 Datenbanken Sequenzformate 2 Multiple 3 FASTA BLAST 4 Datenbanken Überblick Datenbanken Sequenzformate exponentielles
Enno Abteilung Theoretische Informatik Universität Ulm October 18, 2016 Überblick 1 Datenbanken Sequenzformate 2 Multiple 3 FASTA BLAST 4 Datenbanken Überblick Datenbanken Sequenzformate exponentielles
Sequenzen-Alignierung in der Bioinformatik
 Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 2. VO 2.0.2006 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Sequenzen-Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS 2. VO 2.0.2006 Literatur für diese VO Volker Heun: Skriptum zur Vorlesung
Alignment-Verfahren zum Vergleich biologischer Sequenzen
 zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten
 1 Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten (inkl. Anleitungen zur Recherche von Sequenzen mit GenBank und zur Analyse mit GeneDoc) In der Computer-basierten Version
1 Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten (inkl. Anleitungen zur Recherche von Sequenzen mit GenBank und zur Analyse mit GeneDoc) In der Computer-basierten Version
Bioinformatik. Distanzbasierte phylogenetische Algorithmen. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Distanzbasierte phylogenetische Algorithmen Ulf Leser Wissensmanagement in der Bioinformatik Phylogenetische Bäume Stammbaum (Phylogenetic Tree) Ulf Leser: Algorithmische Bioinformatik, Wintersemester
Bioinformatik Distanzbasierte phylogenetische Algorithmen Ulf Leser Wissensmanagement in der Bioinformatik Phylogenetische Bäume Stammbaum (Phylogenetic Tree) Ulf Leser: Algorithmische Bioinformatik, Wintersemester
Übungen zur Vorlesung Algorithmische Bioinformatik
 Übungen zur Vorlesung Algorithmische Bioinformatik Freie Universität Berlin, WS 2006/07 Utz J. Pape Johanna Ploog Hannes Luz Martin Vingron Blatt 6 Ausgabe am 27.11.2006 Abgabe am 4.12.2006 vor Beginn
Übungen zur Vorlesung Algorithmische Bioinformatik Freie Universität Berlin, WS 2006/07 Utz J. Pape Johanna Ploog Hannes Luz Martin Vingron Blatt 6 Ausgabe am 27.11.2006 Abgabe am 4.12.2006 vor Beginn
Informationsvisualisierung
 Informationsvisualisierung Thema: 7. Visualisierung Biologischer Daten Dozent: Dr. Dirk Zeckzer zeckzer@informatik.uni-leipzig.de Sprechstunde: nach Vereinbarung Umfang: 2 Prüfungsfach: Modul Fortgeschrittene
Informationsvisualisierung Thema: 7. Visualisierung Biologischer Daten Dozent: Dr. Dirk Zeckzer zeckzer@informatik.uni-leipzig.de Sprechstunde: nach Vereinbarung Umfang: 2 Prüfungsfach: Modul Fortgeschrittene
MM Proteinmodelling. Michael Meyer. Vorlesung XVII
 Proteinmodelling Vorlesung XVII Proteinstrukturen Es besteht ein sehr großer Bedarf an Proteinstrukturen: Die Kenntnis der 3D-Struktur hat große Vorteile für das Design neuer Wirkstoffe. Experimentelle
Proteinmodelling Vorlesung XVII Proteinstrukturen Es besteht ein sehr großer Bedarf an Proteinstrukturen: Die Kenntnis der 3D-Struktur hat große Vorteile für das Design neuer Wirkstoffe. Experimentelle
TreeTOPS. Ein Phylogenetik-Icebreaker Spiel. Lehrer- Handbuch. ELLS Europäisches Lernlabor für die Lebenswissenschaften
 TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
Was ist Bioinformatik?
 9. Kurstag: Bioinformatik Der Begriff "Bioinformatik" wurde 1989 erstmals von D.R. Masys im JOURNAL OF RESEARCH OF THE NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY erwähnt. Was ist Bioinformatik? Die
9. Kurstag: Bioinformatik Der Begriff "Bioinformatik" wurde 1989 erstmals von D.R. Masys im JOURNAL OF RESEARCH OF THE NATIONAL INSTITUTE OF STANDARDS AND TECHNOLOGY erwähnt. Was ist Bioinformatik? Die
Bioinformatik. Methoden zur Vorhersage vo n RNA- und Proteinstrukture n. Gerhard Steger
 Bioinformatik Methoden zur Vorhersage vo n RNA- und Proteinstrukture n Gerhard Steger Vorwort ix Strukturvorhersage von Nukleinsäuren 1 Struktur und Funktion von RNA 3 1.1 RNA-Struktur fl 1.2 Thermodynamik
Bioinformatik Methoden zur Vorhersage vo n RNA- und Proteinstrukture n Gerhard Steger Vorwort ix Strukturvorhersage von Nukleinsäuren 1 Struktur und Funktion von RNA 3 1.1 RNA-Struktur fl 1.2 Thermodynamik
Kapitel 7: Sequenzen- Alignierung in der Bioinformatik
 Kapitel 7: Sequenzen- Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS11 19. VO 14. Juni 2007 1 Literatur für diese VO Volker
Kapitel 7: Sequenzen- Alignierung in der Bioinformatik VO Algorithm Engineering Professor Dr. Petra Mutzel Lehrstuhl für Algorithm Engineering, LS11 19. VO 14. Juni 2007 1 Literatur für diese VO Volker
Ursprung und Evolution der Landpflanzen: Untersuchungen zur molekularen Phylogenie der Charophyceae, Moose und Farngewächse
 Ursprung und Evolution der Landpflanzen: Untersuchungen zur molekularen Phylogenie der Charophyceae, Moose und Farngewächse Den Naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität Erlangen-Nümberg
Ursprung und Evolution der Landpflanzen: Untersuchungen zur molekularen Phylogenie der Charophyceae, Moose und Farngewächse Den Naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität Erlangen-Nümberg
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik. Dynamische Programmierung. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Bioinformatik I (Einführung)
") Kay Diederichs, Sommersemester 2015 Bioinformatik I (Einführung) Algorithmen Sequenzen Strukturen PDFs unter http://strucbio.biologie.unikonstanz.de/~dikay/bioinformatik/ Klausur: Fr 17.7. 10:00-11:00
Kay Diederichs, Sommersemester 2015 Bioinformatik I (Einführung) Algorithmen Sequenzen Strukturen PDFs unter http://strucbio.biologie.unikonstanz.de/~dikay/bioinformatik/ Klausur: Fr 17.7. 10:00-11:00
Bioinformatik: Neue Paradigmen in der Forschung
 Bioinformatik: Neue Paradigmen in der Forschung Sommerakademie der Studienstiftung St. Johann, 1.-14.9.2002 Leitung: Proff. Ernst W. Mayr & Rolf Backofen Thema 4: Paarweises und multiples (heuristisches)
Bioinformatik: Neue Paradigmen in der Forschung Sommerakademie der Studienstiftung St. Johann, 1.-14.9.2002 Leitung: Proff. Ernst W. Mayr & Rolf Backofen Thema 4: Paarweises und multiples (heuristisches)
Einführung in die Bioinformatik
 Einführung in die Bioinformatik SS 2012 1. Was ist Bioinformatik? Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de Ablauf und Formales Ringvorlesung
Einführung in die Bioinformatik SS 2012 1. Was ist Bioinformatik? Kay Nieselt Integrative Transkriptomik Zentrum für Bioinformatik Tübingen Kay.Nieselt@uni-tuebingen.de Ablauf und Formales Ringvorlesung
Effiziente Algorithmen
 Überblick Ausgewählte Algorithmen der Bioinformatik Effiziente Algorithmen Ausgewählte Algorithmen der Bioinformatik: Teil 1 Markus Leitner (Martin Gruber, Gabriele Koller) 1. Einführung in die Molekularbiologie
Überblick Ausgewählte Algorithmen der Bioinformatik Effiziente Algorithmen Ausgewählte Algorithmen der Bioinformatik: Teil 1 Markus Leitner (Martin Gruber, Gabriele Koller) 1. Einführung in die Molekularbiologie
1 Schulinterner Kernlehrplan Biologie Q2 Evolution
 1 Schulinterner Kernlehrplan Biologie Q2 Evolution 1 Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen Evolution Evolutionstheorien LK Evolutionstheorie Biodiversität und Systematik Entwicklung
1 Schulinterner Kernlehrplan Biologie Q2 Evolution 1 Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen Evolution Evolutionstheorien LK Evolutionstheorie Biodiversität und Systematik Entwicklung
Bioinformatik an der FH Bingen
 Bioinformatik an der FH Bingen Prof. Dr. Antje Krause 05.11.2010 Wie alles begann... 1955 erste Proteinsequenz (nach 12 Jahren Arbeit) veröffentlicht (Insulin vom Rind) Frederick Sanger MALWTRLRPLLALLALWPPPPA
Bioinformatik an der FH Bingen Prof. Dr. Antje Krause 05.11.2010 Wie alles begann... 1955 erste Proteinsequenz (nach 12 Jahren Arbeit) veröffentlicht (Insulin vom Rind) Frederick Sanger MALWTRLRPLLALLALWPPPPA
Inhalt. Entdeckung und allgemeine Informationen. Klassifizierung. Genom Viren untypische Gene Tyrosyl-tRNA Synthetase. Ursprung von grossen DNA Viren
 Mimivirus Inhalt Entdeckung und allgemeine Informationen Klassifizierung Genom Viren untypische Gene Tyrosyl-tRNA Synthetase Ursprung von grossen DNA Viren Entstehung von Eukaryoten Entdeckung 1992 in
Mimivirus Inhalt Entdeckung und allgemeine Informationen Klassifizierung Genom Viren untypische Gene Tyrosyl-tRNA Synthetase Ursprung von grossen DNA Viren Entstehung von Eukaryoten Entdeckung 1992 in
Die Suche nach Genen in Bakteriengenomen. BWInf-Workshop 22.-23. März 2011. Prof. Dr. Sven Rahmann AG Bioinformatik Informatik XI, TU Dortmund
 Die Suche nach Genen in Bakteriengenomen BWInf-Workshop 22.-23. März 2011 Prof. Dr. Sven Rahmann AG Bioinformatik Informatik XI, TU Dortmund 1 Bioinformatik was ist das? Aufgabe: Analyse (molekular)biologischer
Die Suche nach Genen in Bakteriengenomen BWInf-Workshop 22.-23. März 2011 Prof. Dr. Sven Rahmann AG Bioinformatik Informatik XI, TU Dortmund 1 Bioinformatik was ist das? Aufgabe: Analyse (molekular)biologischer
Informationstechnologie in der Pflanzenzüchtung. Biocomputing in einem Züchtungsunternehmen. Andreas Menze KWS SAAT AG, Einbeck
 Informationstechnologie in der Pflanzenzüchtung Biocomputing in einem Züchtungsunternehmen Andreas Menze KWS SAAT AG, Einbeck Biocomputing in einem Züchtungsunternehmen Biocomputing Was ist das? Wozu wird
Informationstechnologie in der Pflanzenzüchtung Biocomputing in einem Züchtungsunternehmen Andreas Menze KWS SAAT AG, Einbeck Biocomputing in einem Züchtungsunternehmen Biocomputing Was ist das? Wozu wird
Luke Alphey. DNA-Sequenzierung. Aus dem Englischen übersetzt von Kurt Beginnen. Spektrum Akademischer Verlag
 Luke Alphey DNA-Sequenzierung Aus dem Englischen übersetzt von Kurt Beginnen Spektrum Akademischer Verlag Inhalt Abkürzungen 11 Vorwort 15 Danksagung 16 Teil 1: Grundprinzipien und Methoden 1. 1.1 1.2
Luke Alphey DNA-Sequenzierung Aus dem Englischen übersetzt von Kurt Beginnen Spektrum Akademischer Verlag Inhalt Abkürzungen 11 Vorwort 15 Danksagung 16 Teil 1: Grundprinzipien und Methoden 1. 1.1 1.2
Neue DNA Sequenzierungstechnologien im Überblick
 Neue DNA Sequenzierungstechnologien im Überblick Dr. Bernd Timmermann Next Generation Sequencing Core Facility Max Planck Institute for Molecular Genetics Berlin, Germany Max-Planck-Gesellschaft 80 Institute
Neue DNA Sequenzierungstechnologien im Überblick Dr. Bernd Timmermann Next Generation Sequencing Core Facility Max Planck Institute for Molecular Genetics Berlin, Germany Max-Planck-Gesellschaft 80 Institute
Übung II. Einführung, Teil 1. Arbeiten mit Ensembl
 Übung II Einführung, Teil 1 Arbeiten mit Ensembl Ensembl Genome Browser (Bereitstellung von Vielzeller Genomen) Projekt wurde 1999 initiiert Projektpartner EMBL European Bioinformatics Institute (EBI)
Übung II Einführung, Teil 1 Arbeiten mit Ensembl Ensembl Genome Browser (Bereitstellung von Vielzeller Genomen) Projekt wurde 1999 initiiert Projektpartner EMBL European Bioinformatics Institute (EBI)
27 Funktionelle Genomanalysen Sachverzeichnis
 Inhaltsverzeichnis 27 Funktionelle Genomanalysen... 543 27.1 Einleitung... 543 27.2 RNA-Interferenz: sirna/shrna-screens 543 Gunter Meister 27.3 Knock-out-Technologie: homologe Rekombination im Genom der
Inhaltsverzeichnis 27 Funktionelle Genomanalysen... 543 27.1 Einleitung... 543 27.2 RNA-Interferenz: sirna/shrna-screens 543 Gunter Meister 27.3 Knock-out-Technologie: homologe Rekombination im Genom der
Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut, keine vorgegebenen Klassen
 7. Clusteranalyse (= Häufungsanalyse; Clustering-Verfahren) wird der multivariaten Statistik zugeordnet Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut,
7. Clusteranalyse (= Häufungsanalyse; Clustering-Verfahren) wird der multivariaten Statistik zugeordnet Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut,
Statistische Verfahren:
 Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Personalisierte Medizin
 Personalisierte Medizin Möglichkeiten und Grenzen Prof. Dr. Friedemann Horn Universität Leipzig, Institut für Klinische Immunologie, Molekulare Immunologie Fraunhofer Institut für Zelltherapie und Immunologie
Personalisierte Medizin Möglichkeiten und Grenzen Prof. Dr. Friedemann Horn Universität Leipzig, Institut für Klinische Immunologie, Molekulare Immunologie Fraunhofer Institut für Zelltherapie und Immunologie
Verwandtschaftsbestimmung mit molekularen Daten
 Verwandtschaftsbestimmung mit molekularen Daten DITTMAR GRAF Online-Ergänzung MNU 67/5 (15.7.2014) Seiten 1 6, ISSN 0025-5866, Verlag Klaus Seeberger, Neuss 1 DITTMAR GRAF Verwandtschaftsbestimmung mit
Verwandtschaftsbestimmung mit molekularen Daten DITTMAR GRAF Online-Ergänzung MNU 67/5 (15.7.2014) Seiten 1 6, ISSN 0025-5866, Verlag Klaus Seeberger, Neuss 1 DITTMAR GRAF Verwandtschaftsbestimmung mit
Algorithmen und Datenstrukturen in der Bioinformatik Viertes Übungsblatt WS 05/06 Musterlösung
 Konstantin Clemens Johanna Ploog Freie Universität Berlin Institut für Mathematik II Arbeitsgruppe für Mathematik in den Lebenswissenschaften Algorithmen und Datenstrukturen in der Bioinformatik Viertes
Konstantin Clemens Johanna Ploog Freie Universität Berlin Institut für Mathematik II Arbeitsgruppe für Mathematik in den Lebenswissenschaften Algorithmen und Datenstrukturen in der Bioinformatik Viertes
MOL.504 Analyse von DNA- und Proteinsequenzen. Übungsaufgaben BLAST-Sequenzsuche und -vergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben BLAST-Sequenzsuche und -vergleiche Ü6a blastn und blastx Verwenden Sie die in Übung 3 (Datenbanken) gefundene yqjm-sequenz aus Bacillus subtilis
MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben BLAST-Sequenzsuche und -vergleiche Ü6a blastn und blastx Verwenden Sie die in Übung 3 (Datenbanken) gefundene yqjm-sequenz aus Bacillus subtilis
Abiturprüfung Biologie, Grundkurs
 Seite 1 von 5 Abiturprüfung 2009 Biologie, Grundkurs Aufgabenstellung: Thema: Parasiten als Indikatoren der Primaten-Evolution I.1 Begründen Sie, warum der Vergleich von Aminosäure-Sequenzen und Basen-Sequenzen
Seite 1 von 5 Abiturprüfung 2009 Biologie, Grundkurs Aufgabenstellung: Thema: Parasiten als Indikatoren der Primaten-Evolution I.1 Begründen Sie, warum der Vergleich von Aminosäure-Sequenzen und Basen-Sequenzen
FOR MUW SSM3 (2008) STUDENTS EDUCATIONAL PURPOSE ONLY
 STUDENTS EDUCATIONAL PURPOSE ONLY") Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
Angewandte Bioinformatik Grundlagen der Annotation von eukaryotischen Genomen Online Datenbanken und Bioinformatik Tools Sequenzen Sequenzalignment: Fasta und Blast Motive und Hidden Markov Models Genom-Browser
Biowissenschaftlich recherchieren
 Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
UniZH Fallstudie Mikrobiologie (BIO126) SS2006
 SS2006") Universität Zürich, Fallstudie SS 2006, Wahlmodul Mikro-Biologie, Bio-126, Gruppe 07 Wie gross ist die evolutionäre Distanz zwischen dem von Clostridium tetani gebildeten Tetanustoxin und den sieben von
Universität Zürich, Fallstudie SS 2006, Wahlmodul Mikro-Biologie, Bio-126, Gruppe 07 Wie gross ist die evolutionäre Distanz zwischen dem von Clostridium tetani gebildeten Tetanustoxin und den sieben von
Neutrale Evolution vs. Selektion basierend auf einer Sequenz
 Neutrale Evolution vs. Selektion basierend auf einer Sequenz Ulrich Seyfarth 16. November 2004 Inhaltsverzeichnis 1 Einführung 1 1.1 Geschichte............................... 1 1.2 Motivation...............................
Neutrale Evolution vs. Selektion basierend auf einer Sequenz Ulrich Seyfarth 16. November 2004 Inhaltsverzeichnis 1 Einführung 1 1.1 Geschichte............................... 1 1.2 Motivation...............................
Stefan Rensing erforscht evolutionären Übergang von Algen zu Landpflanzen
 Powered by Seiten-Adresse: https://www.gesundheitsindustriebw.de/de/fachbeitrag/aktuell/stefan-rensing-erforschtevolutionaeren-bergang-von-algen-zu-landpflanzen/ Stefan Rensing erforscht evolutionären
Powered by Seiten-Adresse: https://www.gesundheitsindustriebw.de/de/fachbeitrag/aktuell/stefan-rensing-erforschtevolutionaeren-bergang-von-algen-zu-landpflanzen/ Stefan Rensing erforscht evolutionären
Mathematik in den Life Siences
 Gerhard Keller Mathematik in den Life Siences Grundlagen der Modellbildung und Statistik mit einer Einführung in die Statistik-Software R 49 Abbildungen Verlag Eugen Ulmer Stuttgart Inhaltsverzeichnis
Gerhard Keller Mathematik in den Life Siences Grundlagen der Modellbildung und Statistik mit einer Einführung in die Statistik-Software R 49 Abbildungen Verlag Eugen Ulmer Stuttgart Inhaltsverzeichnis
Effiziente Algorithmen
 Überblick Ausgewählte Algorithmen der Bioinformatik Effiziente Algorithmen Ausgewählte Algorithmen der Bioinformatik: Teil 2 Markus Leitner (Martin Gruber, Gabriele Koller) 1. Einführung in die Molekularbiologie
Überblick Ausgewählte Algorithmen der Bioinformatik Effiziente Algorithmen Ausgewählte Algorithmen der Bioinformatik: Teil 2 Markus Leitner (Martin Gruber, Gabriele Koller) 1. Einführung in die Molekularbiologie
Algorithmen auf Sequenzen 12.04.2010
 Algorithmen auf Sequenzen 12.04.2010 Prof. Dr. Sven Rahmann 1 Team Prof. Dr. Sven Rahmann Dipl.-Inform Tobias Marschall (Skript) Zeit Mo 8:30-10; Übungen Mi 8:30-10 ca. alle 2 Wochen (Plan!) Ort OH14,
Algorithmen auf Sequenzen 12.04.2010 Prof. Dr. Sven Rahmann 1 Team Prof. Dr. Sven Rahmann Dipl.-Inform Tobias Marschall (Skript) Zeit Mo 8:30-10; Übungen Mi 8:30-10 ca. alle 2 Wochen (Plan!) Ort OH14,
Kapitel 12: Schnelles Bestimmen der Frequent Itemsets
 Einleitung In welchen Situationen ist Apriori teuer, und warum? Kapitel 12: Schnelles Bestimmen der Frequent Itemsets Data Warehousing und Mining 1 Data Warehousing und Mining 2 Schnelles Identifizieren
Einleitung In welchen Situationen ist Apriori teuer, und warum? Kapitel 12: Schnelles Bestimmen der Frequent Itemsets Data Warehousing und Mining 1 Data Warehousing und Mining 2 Schnelles Identifizieren
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 16 - Mittwoch. Annkatrin Bressin Freie Universität Berlin
 WS2013/2014 Woche 16 - Mittwoch. Annkatrin Bressin Freie Universität Berlin") VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 16 - Mittwoch Annkatrin Bressin Freie Universität Berlin Vorlesungsthemen Part 1: Background Basics (4) 1. The Nucleic Acid World 2. Protein Structure
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 16 - Mittwoch Annkatrin Bressin Freie Universität Berlin Vorlesungsthemen Part 1: Background Basics (4) 1. The Nucleic Acid World 2. Protein Structure
Ein molekularer Stammbaum der Greifvögel
 Powered by Seiten-Adresse: https://www.biooekonomiebw.de/de/fachbeitrag/aktuell/ein-molekularerstammbaum-der-greifvoegel/ Ein molekularer Stammbaum der Greifvögel An der Universität Heidelberg leitet Prof.
Powered by Seiten-Adresse: https://www.biooekonomiebw.de/de/fachbeitrag/aktuell/ein-molekularerstammbaum-der-greifvoegel/ Ein molekularer Stammbaum der Greifvögel An der Universität Heidelberg leitet Prof.
BLAST Basic Local Alignment Search Tool
 BLAST Basic Local Alignment Search Tool Martin Winkels 21.12.2012 wissen leben WWU Münster Institut für Medizinische Informatik Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis 1 Einleitung 2 2
BLAST Basic Local Alignment Search Tool Martin Winkels 21.12.2012 wissen leben WWU Münster Institut für Medizinische Informatik Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis 1 Einleitung 2 2
Das Human-Genom und seine sich abzeichnende Dynamik
 Das Human-Genom und seine sich abzeichnende Dynamik Matthias Platzer Genomanalyse Leibniz Institut für Altersforschung - Fritz-Lipmann Institut (FLI) Humangenom - Sequenzierung 1. Methode 2. Ergebnisse
Das Human-Genom und seine sich abzeichnende Dynamik Matthias Platzer Genomanalyse Leibniz Institut für Altersforschung - Fritz-Lipmann Institut (FLI) Humangenom - Sequenzierung 1. Methode 2. Ergebnisse
Python im Bioinformatiker-Alltag
 Python im Bioinformatiker-Alltag Marcel Martin Bioinformatik für Hochdurchsatztechnologien, TU Dortmund 6. Oktober 2011 1 von 27 Worum gehts? Bioinformatik Löst Probleme in Biologie oder Medizin Teilbereich
Python im Bioinformatiker-Alltag Marcel Martin Bioinformatik für Hochdurchsatztechnologien, TU Dortmund 6. Oktober 2011 1 von 27 Worum gehts? Bioinformatik Löst Probleme in Biologie oder Medizin Teilbereich
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr
 Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
Übung II. Einführung. Teil 1 Arbeiten mit Sequenzen recombinante DNA
 Übung II Einführung Teil 1 Arbeiten mit Sequenzen recombinante DNA Recombinante DNA Technologie Protein Synthese In vitro Expression Libraries Gene Transfer in Tieren und Pflanzen Recombinante DNA Technologie
Übung II Einführung Teil 1 Arbeiten mit Sequenzen recombinante DNA Recombinante DNA Technologie Protein Synthese In vitro Expression Libraries Gene Transfer in Tieren und Pflanzen Recombinante DNA Technologie
Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio
 Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio Ein Wissenschaftler erhält nach einer Sequenzierung folgenden Ausschnitt aus einer DNA-Sequenz: 5 ctaccatcaa tccggtaggt tttccggctg
Modul 8: Bioinformatik A. Von der DNA zum Protein Proteinsynthese in silicio Ein Wissenschaftler erhält nach einer Sequenzierung folgenden Ausschnitt aus einer DNA-Sequenz: 5 ctaccatcaa tccggtaggt tttccggctg
Algorithmische Bioinformatik
 lgorithmische Bioinformatik Stringalignment Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung pproximative Stringvergleiche Dotplots Edit-bstand und lignment Naiver lgorithmus Ulf
lgorithmische Bioinformatik Stringalignment Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung pproximative Stringvergleiche Dotplots Edit-bstand und lignment Naiver lgorithmus Ulf
3 Bioinformatik I, Einführung, Grundlagen
 Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2009/2010 Hinweis: Dies ist eine stichpunktartige Liste der wichtigen Themen
Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2009/2010 Hinweis: Dies ist eine stichpunktartige Liste der wichtigen Themen
Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST
 Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST 10.06.2010 Prof. Dr. Sven Rahmann 1 Sequenzvergleich: Motivation Hat man die DNA-Sequenz eines Gens, die Aminosäuresequenz
Einführung in die Angewandte Bioinformatik: Sequenzähnlichkeit, Sequenzalignment, BLAST 10.06.2010 Prof. Dr. Sven Rahmann 1 Sequenzvergleich: Motivation Hat man die DNA-Sequenz eines Gens, die Aminosäuresequenz
Klassische und real-time RT-PCR-Teste
 F L I Klassische und real-time RT-PCR-Teste - molekulare Diagnostik aviärer Influenzavirusinfektionen -! "!"#$ % &!'(!!'(! #"#)* +,!"#$!!'(!!'(!-.( /0 * /" * #!$ % & '(!-.(2314415 /'(!2314415 /6'(!2678.5
F L I Klassische und real-time RT-PCR-Teste - molekulare Diagnostik aviärer Influenzavirusinfektionen -! "!"#$ % &!'(!!'(! #"#)* +,!"#$!!'(!!'(!-.( /0 * /" * #!$ % & '(!-.(2314415 /'(!2314415 /6'(!2678.5
Projektskizze: Studie/ Auswertung/ Publikation
 Projektskizze: Studie/ Auswertung/ Publikation Name: PD Dr. Dr. Robert Bals, CAPNetz C11 Datum: 30. 8. 2007 Institution(en)/Autor(en): Klinikum der Universität Marburg Klinik für Innere Medizin mit Schwerpunkt
Projektskizze: Studie/ Auswertung/ Publikation Name: PD Dr. Dr. Robert Bals, CAPNetz C11 Datum: 30. 8. 2007 Institution(en)/Autor(en): Klinikum der Universität Marburg Klinik für Innere Medizin mit Schwerpunkt
Inhalt 1 Modellorganismen
 Inhalt 1 Modellorganismen....................................... 1 1.1 Escherichia coli....................................... 1 1.1.1 Historisches...................................... 3 1.1.2 Lebenszyklus.....................................
Inhalt 1 Modellorganismen....................................... 1 1.1 Escherichia coli....................................... 1 1.1.1 Historisches...................................... 3 1.1.2 Lebenszyklus.....................................
Bei Einbeziehung von neun Allelen in den Vergleich ergibt sich eine Mutation in 38 Generationen (350:9); das entspricht ca. 770 Jahren.
; das entspricht ca. 770 Jahren.") 336 DNA Genealogie Das Muster genetischer Variationen im Erbgut einer Person - zu gleichen Teilen von Mutter und Vater ererbt - definiert seine genetische Identität unveränderlich. Neben seiner Identität
336 DNA Genealogie Das Muster genetischer Variationen im Erbgut einer Person - zu gleichen Teilen von Mutter und Vater ererbt - definiert seine genetische Identität unveränderlich. Neben seiner Identität
Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2016
 Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2016 Vorbemerkung für die Erlangung des Testats: Bearbeiten Sie die unten gestellten Aufgaben
Biologie I/B: Klassische und molekulare Genetik, molekulare Grundlagen der Entwicklung Theoretische Übungen SS 2016 Vorbemerkung für die Erlangung des Testats: Bearbeiten Sie die unten gestellten Aufgaben
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
BACHELORARBEIT. Herr B.Sc Mathias Langer. Analyse der molekularbiologischen Evolution der BRCT- Domäne im Energieprofilorientierten
 BACHELORARBEIT Herr B.Sc Mathias Langer Analyse der molekularbiologischen Evolution der BRCT- Domäne im Energieprofilorientierten Kontext Mittweida, 2012 Fakultät Mathematik/ Naturwissenschaften/ Informatik
BACHELORARBEIT Herr B.Sc Mathias Langer Analyse der molekularbiologischen Evolution der BRCT- Domäne im Energieprofilorientierten Kontext Mittweida, 2012 Fakultät Mathematik/ Naturwissenschaften/ Informatik
Ausgewählte Algorithmen der Bioinformatik
 Überblick Ausgewählte Algorithmen der Bioinformatik 1. Einführung in die Molekularbiologie 2. Vergleich von Sequenzen 2.1 Paarweises Sequence Alignment 2.2 Multiple Sequence Alignment 2.3 Vergleichsmaße:
Überblick Ausgewählte Algorithmen der Bioinformatik 1. Einführung in die Molekularbiologie 2. Vergleich von Sequenzen 2.1 Paarweises Sequence Alignment 2.2 Multiple Sequence Alignment 2.3 Vergleichsmaße:
Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
 Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof Daniel Stöckel, M. Sc. Patrick Trampert, M. Sc. Lara Schneider, M. Sc. I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
Universität des Saarlandes FR 6.2 Informatik Prof. Dr. Hans-Peter Lenhof Daniel Stöckel, M. Sc. Patrick Trampert, M. Sc. Lara Schneider, M. Sc. I Wichtige Themen aus der Vorlesung Bioinformatik I WS 2013/2014
MOL.504 Analyse von DNA- und Proteinsequenzen. Übungsaufgaben Datenbanken und Informationssysteme
 MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben Datenbanken und Informationssysteme Ü1 Tutorial für NCBI NCBI Nucleotide: Suche nach cellobiose dehydrogenase fungi Ü1 Tutorial für NCBI NCBI
MOL.504 Analyse von DNA- und Proteinsequenzen Übungsaufgaben Datenbanken und Informationssysteme Ü1 Tutorial für NCBI NCBI Nucleotide: Suche nach cellobiose dehydrogenase fungi Ü1 Tutorial für NCBI NCBI
Serie 10: Inverse Matrix und Determinante
 D-ERDW, D-HEST, D-USYS Mathematik I HS 5 Dr Ana Cannas Serie 0: Inverse Matrix und Determinante Bemerkung: Die Aufgaben dieser Serie bilden den Fokus der Übungsgruppen vom und 5 November Gegeben sind die
D-ERDW, D-HEST, D-USYS Mathematik I HS 5 Dr Ana Cannas Serie 0: Inverse Matrix und Determinante Bemerkung: Die Aufgaben dieser Serie bilden den Fokus der Übungsgruppen vom und 5 November Gegeben sind die
Übungsblatt Molekularbiologie und Genetik für Studierende der Bioinformatik II 1. Übung
 Übungsblatt Molekularbiologie und Genetik für Studierende der Bioinformatik II 1 Name des Studierenden: Datum: Einführung Übung Bitte bereiten Sie folgende Probleme vor der Übung am Mittwoch. Die Klausur
Übungsblatt Molekularbiologie und Genetik für Studierende der Bioinformatik II 1 Name des Studierenden: Datum: Einführung Übung Bitte bereiten Sie folgende Probleme vor der Übung am Mittwoch. Die Klausur
Bioinformatik Statistik und Analyse mit R 22.05.2009-1 -
 Bioinformatik Statistik und Analyse mit R 22.05.2009-1 - Definition: Bioinformatik Die Bioinformatik http://de.wikipedia.org/wiki/bioinformatik (englisch bioinformatics, auch computational biology) ist
Bioinformatik Statistik und Analyse mit R 22.05.2009-1 - Definition: Bioinformatik Die Bioinformatik http://de.wikipedia.org/wiki/bioinformatik (englisch bioinformatics, auch computational biology) ist
Wo waren wir stehen geblieben? Evolutions modelle
 Wo waren wir stehen geblieben? Evolutions modelle 1 Stammbaumerstellung 1. Distanz-orientierte Methoden UPGMA (Unweighted Pair-Group Method with Arithmetric Means) Neighbor-joining Minimal Evolution =>
Wo waren wir stehen geblieben? Evolutions modelle 1 Stammbaumerstellung 1. Distanz-orientierte Methoden UPGMA (Unweighted Pair-Group Method with Arithmetric Means) Neighbor-joining Minimal Evolution =>
Organisation und Evolution des Genoms
 Organisation und Evolution des Genoms Organisation und Evolution des Genoms Definition Genom: vollständige DNA-Sequenz eines Organismus I. Einfachstes Genom: Prokaryoten Zwei Gruppen, evolutionär unterschiedlicher
Organisation und Evolution des Genoms Organisation und Evolution des Genoms Definition Genom: vollständige DNA-Sequenz eines Organismus I. Einfachstes Genom: Prokaryoten Zwei Gruppen, evolutionär unterschiedlicher
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Molekulare Arterkennung und Erfassung der Diversität von Engerlingen in der Himalaya Hindukusch-Region
 Molekulare Arterkennung und Erfassung der Diversität von Engerlingen in der Himalaya Hindukusch-Region Corinna Wallinger, Daniela Sint, Bettina Thalinger, Johannes Oehm & Michael Traugott Universität Innsbruck,
Molekulare Arterkennung und Erfassung der Diversität von Engerlingen in der Himalaya Hindukusch-Region Corinna Wallinger, Daniela Sint, Bettina Thalinger, Johannes Oehm & Michael Traugott Universität Innsbruck,
M a s t e r a r b e i t
 Universität Konstanz, FB Informatik und Informationswissenschaft Master-Studiengang Information Engineering M a s t e r a r b e i t Datenanalyse in der Molekularbiologie: Suche nach Abhängigkeiten in alignierten
Universität Konstanz, FB Informatik und Informationswissenschaft Master-Studiengang Information Engineering M a s t e r a r b e i t Datenanalyse in der Molekularbiologie: Suche nach Abhängigkeiten in alignierten