Welche Alignmentmethoden haben Sie bisher kennengelernt?
|
|
|
- Johannes Rothbauer
- vor 6 Jahren
- Abrufe
Transkript

1


2


3 Welche Alignmentmethoden haben Sie bisher kennengelernt?
4 Was heißt optimal? Optimal = die wenigsten Mutationen. Sequenzen bestehen aus Elementen (z.b. Aminosäuren oder Nukleotide).
5 Edit Distanzen sind in Substitutionsmatrizen abgelegt. Hier ist der einfachste Fall abgebildet. Ähnlichkeitsmatrix = je ähnlicher, desto höher der Score, Distanzmatrix = je ähnlicher, desto niedriger der Score. Bsp. Einheitsmatrix Der Algorithmus versucht die Summe der einzelnen Scores aus einer Ähnlichkeitsmatrix zu maximieren. Für Aminosäuren gibt es die PAM (abgeleitet von einem globalen Alignment) und BLOSUM (abgeleitet von einem lokalen Alignment) Matrizen, welche Ähnlichkeitsmatrizen sind. PAMn vs BLOSUMn: PAM-Matrizen mit kleinesrem n sollten bei weniger divergenten Sequenzen genutzt werden. Bei den BLOSUM-Matrizen ist es andersherum.
6 Hamming Distanz kann nur gleichlange Sequenzen vergleichen. Natürlich können aber auch Insertionen und Deletionen auftreten. Insertionen in der einen Sequenz entsprechen Deletionen in der anderen Sequenz. Daher nennt man sie auch übergeordnet Indels.
7 Was ist in unserem Fall die Substitutionsmatrix? Eine Distanzmatrix. Insertion / Deletion in Bezug zur Sequenz auf der horizontalen Achse.

8 Abstand der beiden Sequenzen ist 2.
9 Gehe von rechts nach links entlang der Pfeile und halte die Summe der D i,j minimal. Dann folge der Spur von links oben nach rechts unten. Wenn zwei Einträge horizontal verbunden sind, so muss in der vertikalen Sequenz ein Gap stehen (Insertion in Bezug auf die horizontale Achse). Wenn zwei Einträge vertikal verbunden sind, so muss in der horizontalen Sequenz ein Gap stehen (Deletion in Bezug auf die horizontale Achse).
10 Substitutionsmatrizen können auch beispielsweise Transitionen anders gewichten als Transversionen. Transitionen gibt es theoretisch weniger, treten aber wegen der chemischen Eigenschaften der Nukleinsäuren häufiger auf. Score-basierte Algorithmen maximieren die Punktezahl, was equivalent zum Minimieren der Edit Distance ist. Was ist in unserem Fall die Substitutionsmatrix? Eine Ähnlichkeitsmatrix.
11 Globales Alignment kann zu großen Gaps führen. Globale Alignments werden hauptsächlich verwendet, wenn die zu untersuchenden Sequenzen ähnlich lang sind und starke Sequenzhomologien erwartet werden. Lokalen Alignments werden beispielsweise genutzt, wenn man nach gleichen Sequenzmotiven oder Domänen bei Proteinen sucht. Beide Algorithmen finden immer das optimale Alignment ( Dynamic Programming ). Multiple Alignments finden nicht immer das optimale Alignment und es Bedarf der Kontrolle. Sie erzeugen so genannte Guide Trees, welche die ähnlichsten Sequenzen zusammengruppieren. Häufig wird eine Neighbour-Joining Methode angewendet, um den Baum zu erstellen.
12
13

14 Score des Alignments = -8.
15 Beachte die unterschiedliche Initialisierung. Negative Scores sind nicht möglich. Traceback funktioniert anders (beginne bei der höchsten Zahl und ende, sobald die Null erreicht ist).
16
17

.")
18 Die Konsensussequenz fasst das multiple Alignment so zusammen, dass nur die an jeder Position häufigste Base dargestellt wird. Starke Reduktion der Information. Das Sequenzlogo ist eine graphische Darstellung des Grades der Konserviertheit eines Nukleotids. Die relative Größe der Buchstaben entspricht den Frequenzen der Nukleotide an einer bestimmten Position des Alignments und die absolute Größe der Buchstaben entspricht dem Informationsgehalt an dieser Position (in Bits). Das heißt, je größer die Buchstaben, desto stärker ist diese Position konserviert.


19 Das gleiche Prinzip wird auch bei der Visualisierung von Proteindomänen genutzt.
20


21
22 Die phylogenetische Systematik ist eine Systematik, welche die tatsächliche evolutionäre Beziehung der Organismen zueinander widerspiegelt. Plesiomorphe Merkmale sind ursprüngliche Merkmale, apomorphe Merkmale sind abgeleitete Merkmale. Symplesiomorphe Merkmale sind plesiomorphe Merkmale, welche zwischen Taxa geteilt sind. Autapomorphe Merkmale sind apomorphe Merkmale, welche spezifisch für ein Taxon sind. Synapomorphe Merkmale sind apomorphe Merkmale, welche Taxa gemeinsam haben. Homoplasien sind Merkmale, welche unabhängig in unterschiedlichen Taxa entstanden sind und daher nicht auf eine gemeinsame Abstammung zurückgehen (Konvergenz). In der molekularen Phylogenetik eher Homoplasie genannt. Dies kann durch Rückmutationen oder analoge Mutationen entstehen. Durch unerkannte Homoplasien kann es zur Bildung von polyphyletischen Gruppen kommen. Welche Arten von Merkmalen sind besonders wichtig für die Bildung monophyletischer Gruppen? Synapomorphe Merkmale. Symplesiomorphe Merkmale helfen nicht paraphyletische Gruppen in monophyletische Gruppen aufzuspalten, sie sind aber hilfreich für die evolutionäre Eingruppierung. Was sind paraphyletische Gruppen? Taxa, die zwar auf einen gemeinsamen Vorfahren zurückgehen, aber aus denen auch andere Lebensformen hervorgegangen sind.
, Brückenechsen (Sphenodontia), Echsen und Schlangen (Squamata), Krokodile (Crocodylia).")
23 Ectothermie der Krokodile und Schildkröten: Beide sind ectotherm; dies hilft uns aber nicht dabei, ihre Verwandtschaft aufzuklären. Feder der Vögel. Diapsider Schädel der Vögel (Aves), Brückenechsen (Sphenodontia), Echsen und Schlangen (Squamata), Krokodile (Crocodylia). Homoplasie = Konvergenz des Vertebraten-Flügels.
24 Im besten Fall sind Bäume dichotom, manchmal treten aber auch Polytomien auf. Externer Knoten mit nur einem Nachbar, interner Knoten mit zwei Nachbarn. Externer Knoten = OTUs (Operational taxonomic unit) Unbewurzelte Bäume haben keine richtige Leserichtung. Erst durch die Wurzel lassen sich ältere von jüngeren Verzweigungen unterscheiden. Mittelpunktbewurzelung (Midpoint rooting): Die Wurzel wird in der Mitte der am weitesten entfernten Taxa gesetzt.
25 Besser: Wurzelung geschieht durch eine Außengruppe (Outgroup). Diese ist ein Taxon, das mit Sicherheit stammesgeschichtlich weiter von der Innengruppe entfernt steht, als alle Taxa der Innengruppe zueinander.
26 Kladogramm: Die Länge der terminalen und internen Zweige hat keine Bedeutung, nur die Topologie ist entscheidend. Dies läßt sich auch als Netzwerk darstellen (mit sieben Möglichkeiten zur Bewurzelung, Pfeile). Phylogramm: Der Grad der Verwandtschaft wird quantitativ wiedergegeben durch die unterschiedlichen Längen der horizontalen Äste. Quantitativ = Anzahl der beobachteten Merkmalsaustausche. Dendrogramm: Ultrametrischer Stammbaum, in dem alle Taxa den gleichen Abstand zur Wurzel haben. D.h. man nimmt eine konstante Veränderungsrate an (molekulare Uhr).
27 Das Newick-Format ist ein Computer-lesbares Format, Bäume darzustellen. Schwestergruppen werden dabei in sukzessive verschachtelte, runde Klammern gesetzt und durch Kommata getrennt. Die Baumbeschreibung wird durch ein Semikolon abgeschlossen. Stammbäume lassen sich um die Knoten beliebig drehen (die Topologie bleibt erhalten).
28 In Phylogrammen wird die Länge der Äste durch einen Doppelpunkt abgetrennt nach jedem internen und externen Knoten angegeben.
29 Mindestens 4 Taxa werden benötigt, um unterschiedliche Bäume zu erhalten.
30 Mindestens 3 Taxa werden benötigt, um unterschiedliche Bäume zu erhalten. Jeder der drei ungewurzelten Bäume kann an seinen 5 Ästen gewurzelt werden 3 * 5 Bäume.
31 Die Anzahl möglicher Bäume wächst extrem schnell.
32
33
34 Warum beschreibt die DNA nicht alles? Ortholog: Gemeinsamer Ursprung durch Artbildung Paralog: Gemeinsamer Ursprung durch Genduplikation Falsche Verwandschaft auf Grund einer Vermischung von Paralogen und Orthologen.
35
36 Nur die Synapomorphien enthalten Informationen zu dem zugrundeliegenden Baum.
37 1. Berechne die paarweisen Distanzen in einer Distanzmatrix
38 Wie kommen wir vom Kladogramm zum Phylogramm/Dendrogramm?
39 UPGMA nimmt eine molekulare Uhr an und berechnet so eine ultrametrische Distanz. Dies kann, muss aber natürlich nicht erfüllt sein. Daher ist UPGMA sehr schnell und effizient, gilt aber heute eher als veraltet. Eine weit verbreitete Distanzmethode ist Neighbour-Joining, welches den Baum mit der kürzesten Summe der Astlängen sucht. Substitutionsmodelle berücksichtigen beispielsweise unterschiedliche Änderungsraten von Transitionen und Transversionen oder von synonymen und nicht-synonymen Substitutionen. Für Aminosäuren sind diese Substitutionsmodelle in den PAM und BLOSUM Matrizen zusammengefasst. Nachteile: Durch die Übertragung in Distanzen können unterschiedliche Sequenzen zur gleichen Distanz führen (im Beispiel Distanz in beiden Fällen = 10). Daher lassen sich Distanzen auch nicht wieder in Sequenzen zurückübertragen. Distanzmethoden betrachten nur Ähnlichkeit, nicht die evolutionäre Geschichte. Außerdem lassen sich morphologische und molekulare Merkmale nicht kombinieren. Die phylogenetische Distanz wird unterschätzt, wenn man einfach die Anzahl der Unterschiede zählt (unkorrigierte p-distanz), da es mehrfache Substitutionen einer Base (multiple Hits) geben kann.
40 Um die Astlängen zu erhalten, kann man den UPGMA-Algorithmus anwenden: 1. Berechne alle paarweisen Distanzen 2. Trage alle Werte in eine symmetrische Distanzmatrix D = d ij ein 3. Suche die beiden Sequenzen/Taxa i und j mit der geringsten Distanz d ij und erstelle ein neues Cluster aus c aus beiden 4. Entferne die Taxa i und j aus dem Set aller Taxa 5. Füge das neue Cluster c in die Distanzmatrix ein 6. Berechne die Distanz zwischen dem neuen Cluster c und allen anderen Gruppen als d ck = ( i * d ik + j * d jk ) / ( i + j ) 7. Gehe zu 3. i und j bezeichnen die Kardinalität der Cluster i und j (also die Anzahl der Elemente im jeweiligen Cluster)
41
42
43
44
45 UPGMA liefert ein Dendrogramm.

46 Diskrete Merkmale = DNA- oder Aminosäuren-Sequenzen.
47 Vorteile: - Einfaches, intuitives Prinzip - Für morphologische Daten ist keine andere Methode etabliert Nachteile: - Multiple Hits werden nicht berücksichtigt (Homoplasien bei stark divergenten Sequenzen) - Nicht alle Bäume können analysiert werden bei großen Datensätzen, sehr zeitaufwändig
48
49
50 Diese Position ist nicht parsimonie-informativ.
51 Diese Position ist nicht parsimonie-informativ, da sie fixiert ist.
52
53 Dies wird erreicht durch die Benutzung von Substitutionsmatrizen (zb PAM oder BLOSUM bei Aminosäuren). Ähnliche zur Parsimonie, aber nutzt komplexere Substitutionsmodelle. Vorteile: - Realistischer als Parsimonie - Parsimonie-uniformative Positionen können unter ML informativ sein, da ML berücksichtigt, dass eine Substitution entlang eines langen Zweiges wahrscheinlicher ist als entlang eines kurzen. Autapomorphien treten eher auf langen Zweigen auf. Nachteile: - Sehr rechenintensiv

54
55
56
57 Ohne Gaps gibt es für jede Zeile und für jede Spalte 17 weitere Bäume, d.h. 20 * 20 = 400 Rekonstruktionen. Das gleiche gilt für die zwei anderen Topologien, also 3 * 400 = 1200 Rekonstruktionen für das erste Merkmal. Es gibt 11 Merkmale, also insgesamt 1200 * 11 = Rekonstruktionen.
58 In diesem Beispiel nutzen wir die Scores aus der PAM250-Matrix. Diese Likelihood-Scores werden für jedes Merkmal, jede Rekonstruktion und jede Topologie gerechnet. Natürlich müssen die Scores zuerst in Wahrscheinlichkeiten umgerechnet werden.
59 Diese parsimonie-uniformative Position kann informativ bei der ML-Methode werden.
60 Am Ende ist die Topologie der drei möglichen Topologien optimal, welche die größte Likelihood aufweist.
61 Die Prior Probabilities sind die Wahrscheinlichkeiten der Hypothese, bevor man die Daten angeschaut hat. Markov-Kette: Wandert durch den Raum aller Topologien und nutzt die Information des vorherigen Baums, um entweder in einen neuen Zustand (Baum) zu wechseln oder beim alte zu bleiben. Die Entscheidung über Wechsel oder Verbleiben geschieht über die Likelihood. Nachteil: Abhängig vom Prior
62 Eigentlich sind biologische Replikate ein probates Mittel. Dies kann allerdings in der Phylogenetik schwierig sein. Dann kann man den Bootstrap nutzen. Je mehr Replikate man macht (also je größer das k), desto kleiner wird der Fehler (k=10000 ist eine recht verläßliche Zahl).
63 Manche Merkmale werden genau einmal gezogen.
64 Manche Merkmale gar nicht.
65 Manche Merkmale werden mehr als einmal gezogen.
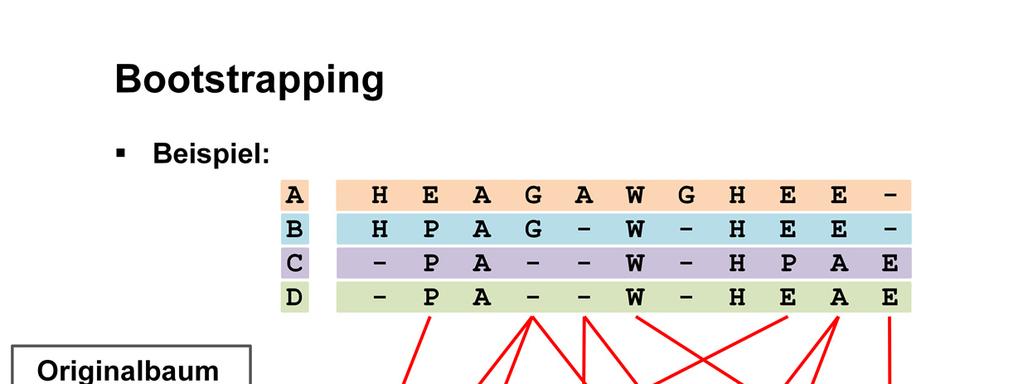
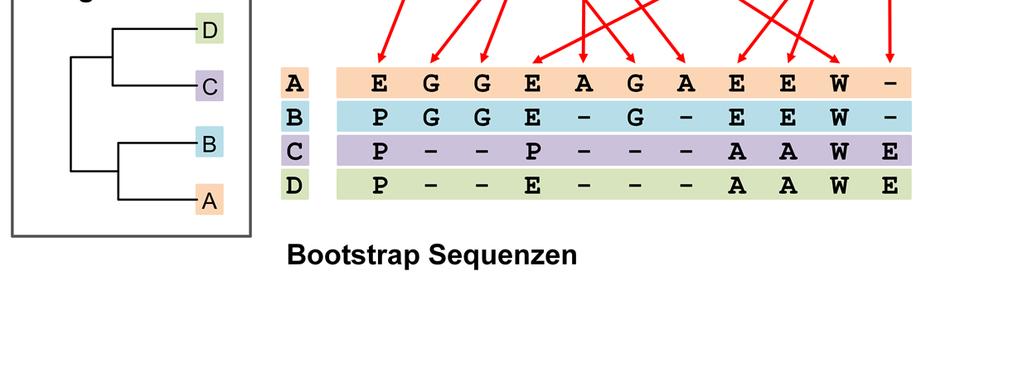
66
67 Anhand der Bootstrap Sequenzen wird ein Baum konstruiert.
68
69 Bootstrap-Wert = 6 (oder 60%).
70 Bootstrap-Wert = 7 (oder 70%). Erfahrungsgemäß sind Bootstrap-Werte von über 70% akzeptabel. Man erzeuge einen Bootstrap-Baum, indem man den Majority Rule Konsensusbaum errechnet. Wenn in einem Replikat mehrere Bäume gleichwahrscheinlich sind, kann man entweder erst einen Strict Consensus Baum innerhalb des Replikats errechnen oder die Bäume im Bootstrap-Baum niedriger gewichten (Frequency-Within-Replicates Ansatz, FWR).
Phylogenetik. Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann. Webseite zur Vorlesung
 Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
Phylogenetik Vorlesung Einführung in die Angewandte Bioinformatik Prof. Dr. Sven Rahmann Webseite zur Vorlesung http://bioinfo.wikidot.com/ Sprechstunde Mo 16-17 in OH14, R214 Sven.Rahmann -at- tu-dortmund.de
TreeTOPS. Ein Phylogenetik-Icebreaker Spiel. Lehrer- Handbuch. ELLS Europäisches Lernlabor für die Lebenswissenschaften
 TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
TreeTOPS Ein Phylogenetik-Icebreaker Spiel Lehrer- Handbuch ELLS Europäisches Lernlabor für die Lebenswissenschaften 1 Übergeordnetes Ziel Das übergeordnete Ziel des Spieles ist es, die Spieler in das
MOL.504 Analyse von DNA- und Proteinsequenzen. Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche
 MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
MOL.504 Analyse von DNA- und Proteinsequenzen Modul 2 BLAST-Sequenzsuche und Sequenzvergleiche Summary Modul 1 - Datenbanken Wo finde ich die DNA Sequenz meines Zielgens? Wie erhalte ich Info aus der DNA-Datenbank
Homologie und Sequenzähnlichkeit. Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Homologie und Sequenzähnlichkeit Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de Homologie Verwandtschaft aufgrund gleicher Abstammung basiert auf Speziation (Artbildung): aus einer
Alignment von DNA- und Proteinsequenzen
 WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
WS2012/2013 Genomforschung und Sequenzanalyse - Einführung in Methoden der Bioinformatik- Thomas Hankeln Alignment von DNA- und Proteinsequenzen das vielleicht wichtigste Werkzeug der Bioinformatik! 1
Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld. Institut für Zoologie Universität Mainz
 Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) Daten einen Stammbaum, und was sagt mir
Fernstudium "Molekulare Phylogenie" Bernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) Daten einen Stammbaum, und was sagt mir
Fernstudium "Molekulare Evolution" Bernhard Lieb Michael Schaffeld. Institut für Zoologie Universität Mainz
 Fernstudium "Molekulare Evolution" ernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) aten einen Stammbaum, und was sagt mir
Fernstudium "Molekulare Evolution" ernhard Lieb Michael Schaffeld Institut für Zoologie Universität Mainz 1 Ziel des Kurses Wie erhalte ich aus meinen (Sequenz-) aten einen Stammbaum, und was sagt mir
Rekonstruktion der Phylogenese
 Rekonstruktion der Phylogenese 15.12.2010 1 Outline Probleme bei der Rekonstruktion eines Stammbaumes: Welche Merkmale? Welche Methode zur Auswahl des wahrscheinlichsten Stammbaumes? Schulen der Klassifikation
Rekonstruktion der Phylogenese 15.12.2010 1 Outline Probleme bei der Rekonstruktion eines Stammbaumes: Welche Merkmale? Welche Methode zur Auswahl des wahrscheinlichsten Stammbaumes? Schulen der Klassifikation
Alignment-Verfahren zum Vergleich biologischer Sequenzen
 zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
zum Vergleich biologischer Sequenzen Hans-Joachim Böckenhauer Dennis Komm Volkshochschule Zürich. April Ein biologisches Problem Fragestellung Finde eine Methode zum Vergleich von DNA-Molekülen oder Proteinen
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de
 Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de") Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Evolution & Genetik (Beispiel Hämoglobin) Prof. Dr. Antje Krause FH Bingen 06721 / 409 253 akrause@fh-bingen.de DNA (Desoxyribonukleinsäure) 5 3 CGATGTACATCG GCTACATGTAGC 3 5 Doppelhelix Basen: Adenin,
Molekulare Phylogenie
 Molekulare Phylogenie Grundbegriffe Methoden der Stammbaum-Rekonstruktion Thomas Hankeln, Institut für Molekulargenetik SS 2010 Grundlagen der molekularen Phylogenie Evolution äußert sich durch Veränderungen
Molekulare Phylogenie Grundbegriffe Methoden der Stammbaum-Rekonstruktion Thomas Hankeln, Institut für Molekulargenetik SS 2010 Grundlagen der molekularen Phylogenie Evolution äußert sich durch Veränderungen
Übungen zur Vorlesung Algorithmische Bioinformatik
 Übungen zur Vorlesung Algorithmische Bioinformatik Freie Universität Berlin, WS 2006/07 Utz J. Pape Johanna Ploog Hannes Luz Martin Vingron Blatt 6 Ausgabe am 27.11.2006 Abgabe am 4.12.2006 vor Beginn
Übungen zur Vorlesung Algorithmische Bioinformatik Freie Universität Berlin, WS 2006/07 Utz J. Pape Johanna Ploog Hannes Luz Martin Vingron Blatt 6 Ausgabe am 27.11.2006 Abgabe am 4.12.2006 vor Beginn
Phylogenetische Analyse
 Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Bioinformatik I - Uebung Phylogenetische Analyse Wenn nicht anders angegeben verwende die Standard-Einstellungen der Programme Hintergrund: Die Schwämme (Phylum Porifera) gehören zu den den ältesten lebenden
Standardbasierter, kompetenzorientierter Unterricht ZPG Biologie 2011 Bildungsplan 2004 Baden-Württemberg Sekundarstufe II - Evolution
 Wie sich die Systematik unter dem lickwinkel der eszendenztheorie verändert ie Systematik ist ein Fachgebiet der iologie. In der Systematik werden Organismen klassifiziert, indem sie in ihrer Vielfalt
Wie sich die Systematik unter dem lickwinkel der eszendenztheorie verändert ie Systematik ist ein Fachgebiet der iologie. In der Systematik werden Organismen klassifiziert, indem sie in ihrer Vielfalt
Lage- und Streuungsparameter
 Lage- und Streuungsparameter Beziehen sich auf die Verteilung der Ausprägungen von intervall- und ratio-skalierten Variablen Versuchen, diese Verteilung durch Zahlen zu beschreiben, statt sie graphisch
Lage- und Streuungsparameter Beziehen sich auf die Verteilung der Ausprägungen von intervall- und ratio-skalierten Variablen Versuchen, diese Verteilung durch Zahlen zu beschreiben, statt sie graphisch
Algorithmische Bioinformatik
 Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
Algorithmische Bioinformatik Effiziente Berechnung des Editabstands Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Inhalt dieser Vorlesung Rekursive Definition des Editabstands
Clustering Seminar für Statistik
 Clustering Markus Kalisch 03.12.2014 1 Ziel von Clustering Finde Gruppen, sodas Elemente innerhalb der gleichen Gruppe möglichst ähnlich sind und Elemente von verschiedenen Gruppen möglichst verschieden
Clustering Markus Kalisch 03.12.2014 1 Ziel von Clustering Finde Gruppen, sodas Elemente innerhalb der gleichen Gruppe möglichst ähnlich sind und Elemente von verschiedenen Gruppen möglichst verschieden
Systematik der Metazoa Eine phylogenetische Übersicht. Version 2.0. Vorwort
 Systematik der Metazoa Eine phylogenetische Übersicht Version 2.0 Vorwort Angeregt durch das Buch Geschichten vom Ursprung des Lebens. Eine Zeitreise auf Darwins Spuren von Richard Dawkins (2009) nahm
Systematik der Metazoa Eine phylogenetische Übersicht Version 2.0 Vorwort Angeregt durch das Buch Geschichten vom Ursprung des Lebens. Eine Zeitreise auf Darwins Spuren von Richard Dawkins (2009) nahm
2. Repräsentationen von Graphen in Computern
 2. Repräsentationen von Graphen in Computern Kapitelinhalt 2. Repräsentationen von Graphen in Computern Matrizen- und Listendarstellung von Graphen Berechnung der Anzahl der verschiedenen Kantenzüge zwischen
2. Repräsentationen von Graphen in Computern Kapitelinhalt 2. Repräsentationen von Graphen in Computern Matrizen- und Listendarstellung von Graphen Berechnung der Anzahl der verschiedenen Kantenzüge zwischen
Einführung in die evolutionäre Bioinformatik Alignmentalgorithmen, Profile, Phylogenetische Analysen
 www.bachelor-and-more.de Einführung in die evolutionäre ioinformatik lignmentalgorithmen, Profile, Phylogenetische nalysen Evolutionsbiologie II für achelor-/lehramtsstudierende 16. Februar 216 Sonja Grath
www.bachelor-and-more.de Einführung in die evolutionäre ioinformatik lignmentalgorithmen, Profile, Phylogenetische nalysen Evolutionsbiologie II für achelor-/lehramtsstudierende 16. Februar 216 Sonja Grath
Informationstheorie als quantitative Methode in der Dialektometrie
 Informationstheorie als quantitative Methode in der Dialektometrie 1 Informationstheorie als quantitative Methode in der Dialektometrie Informationstheorie als quantitative Methode in der Dialektometrie
Informationstheorie als quantitative Methode in der Dialektometrie 1 Informationstheorie als quantitative Methode in der Dialektometrie Informationstheorie als quantitative Methode in der Dialektometrie
Entscheidungsbäume. Definition Entscheidungsbaum. Frage: Gibt es einen Sortieralgorithmus mit o(n log n) Vergleichen?
 Vergleichen?") Entscheidungsbäume Frage: Gibt es einen Sortieralgorithmus mit o(n log n) Vergleichen? Definition Entscheidungsbaum Sei T ein Binärbaum und A = {a 1,..., a n } eine zu sortierenden Menge. T ist ein Entscheidungsbaum
Entscheidungsbäume Frage: Gibt es einen Sortieralgorithmus mit o(n log n) Vergleichen? Definition Entscheidungsbaum Sei T ein Binärbaum und A = {a 1,..., a n } eine zu sortierenden Menge. T ist ein Entscheidungsbaum
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag
 WS2013/2014 Woche 3 - Montag") VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
VL Algorithmische BioInformatik (19710) WS2013/2014 Woche 3 - Montag Tim Conrad AG Medical Bioinformatics Institut für Mathematik & Informatik, Freie Universität Berlin Vorlesungsthemen Part 1: Background
Aufgabensammlung aus Mathematik 2 UMIT, SS 2010, Version vom 7. Mai 2010
 Aufgabensammlung aus Mathematik 2 UMIT, SS 2, Version vom 7. Mai 2 I Aufgabe I Teschl / K 3 Zerlegen Sie die Zahl 8 N in ihre Primfaktoren. Aufgabe II Teschl / K 3 Gegeben sind die natürliche Zahl 7 und
Aufgabensammlung aus Mathematik 2 UMIT, SS 2, Version vom 7. Mai 2 I Aufgabe I Teschl / K 3 Zerlegen Sie die Zahl 8 N in ihre Primfaktoren. Aufgabe II Teschl / K 3 Gegeben sind die natürliche Zahl 7 und
Kürzeste Wege in Graphen. Maurice Duvigneau Otto-von-Guericke Universität Fakultät für Informatik
 Kürzeste Wege in Graphen Maurice Duvigneau Otto-von-Guericke Universität Fakultät für Informatik Gliederung Einleitung Definitionen Algorithmus von Dijkstra Bellmann-Ford Algorithmus Floyd-Warshall Algorithmus
Kürzeste Wege in Graphen Maurice Duvigneau Otto-von-Guericke Universität Fakultät für Informatik Gliederung Einleitung Definitionen Algorithmus von Dijkstra Bellmann-Ford Algorithmus Floyd-Warshall Algorithmus
Bioinformatik für Lebenswissenschaftler
 Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Bioinformatik für Lebenswissenschaftler Oliver Kohlbacher, Steffen Schmidt SS 2010 09. Multiples Alignment I Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Übersicht
Kodierungsalgorithmen
 Kodierungsalgorithmen Komprimierung Verschlüsselung Komprimierung Zielsetzung: Reduktion der Speicherkapazität Schnellere Übertragung Prinzipien: Wiederholungen in den Eingabedaten kompakter speichern
Kodierungsalgorithmen Komprimierung Verschlüsselung Komprimierung Zielsetzung: Reduktion der Speicherkapazität Schnellere Übertragung Prinzipien: Wiederholungen in den Eingabedaten kompakter speichern
3 Quellencodierung. 3.1 Einleitung
 Source coding is what Alice uses to save money on her telephone bills. It is usually used for data compression, in other words, to make messages shorter. John Gordon 3 Quellencodierung 3. Einleitung Im
Source coding is what Alice uses to save money on her telephone bills. It is usually used for data compression, in other words, to make messages shorter. John Gordon 3 Quellencodierung 3. Einleitung Im
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse
 Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
Einführung in die Bioinformatik Algorithmen zur Sequenzanalyse!! Vorläufige Fassung, nur einzelne Abschnitte!!!! Enthält wahrscheinlich noch viele Fehler!!!! Wird regelmäßig erweitert und verbessert!!
Routing Algorithmen. Begriffe, Definitionen
 Begriffe, Definitionen Routing (aus der Informatik) Wegewahl oder Verkehrslenkung bezeichnet in der Telekommunikation das Festlegen von Wegen für Nachrichtenströme bei der Nachrichtenübermittlung über
Begriffe, Definitionen Routing (aus der Informatik) Wegewahl oder Verkehrslenkung bezeichnet in der Telekommunikation das Festlegen von Wegen für Nachrichtenströme bei der Nachrichtenübermittlung über
Domain-independent. independent Duplicate Detection. Vortrag von Marko Pilop & Jens Kleine. SE Data Cleansing
 SE Data Cleansing Domain-independent independent Duplicate Detection Vortrag von Marko Pilop & Jens Kleine http://www.informatik.hu-berlin.de/~pilop/didd.pdf {pilop jkleine}@informatik.hu-berlin.de 1.0
SE Data Cleansing Domain-independent independent Duplicate Detection Vortrag von Marko Pilop & Jens Kleine http://www.informatik.hu-berlin.de/~pilop/didd.pdf {pilop jkleine}@informatik.hu-berlin.de 1.0
5 Zwei spieltheoretische Aspekte
 5 Zwei spieltheoretische Aspekte In diesem Kapitel wollen wir uns mit dem algorithmischen Problem beschäftigen, sogenannte Und-Oder-Bäume (kurz UOB) auszuwerten. Sie sind ein Spezialfall von Spielbäumen,
5 Zwei spieltheoretische Aspekte In diesem Kapitel wollen wir uns mit dem algorithmischen Problem beschäftigen, sogenannte Und-Oder-Bäume (kurz UOB) auszuwerten. Sie sind ein Spezialfall von Spielbäumen,
Wo waren wir stehen geblieben? Evolutions modelle
 Wo waren wir stehen geblieben? Evolutions modelle 1 Stammbaumerstellung 1. Distanz-orientierte Methoden UPGMA (Unweighted Pair-Group Method with Arithmetric Means) Neighbor-joining Minimal Evolution =>
Wo waren wir stehen geblieben? Evolutions modelle 1 Stammbaumerstellung 1. Distanz-orientierte Methoden UPGMA (Unweighted Pair-Group Method with Arithmetric Means) Neighbor-joining Minimal Evolution =>
Approximation in Batch and Multiprocessor Scheduling
 Approximation in Batch and Multiprocessor Scheduling Tim Nonner IBM Research Albert-Ludwigs-Universität Freiburg 3. Dezember 2010 Scheduling Zeit als Ressource und Beschränkung Formaler Gegeben sind Jobs
Approximation in Batch and Multiprocessor Scheduling Tim Nonner IBM Research Albert-Ludwigs-Universität Freiburg 3. Dezember 2010 Scheduling Zeit als Ressource und Beschränkung Formaler Gegeben sind Jobs
BLAST. Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02
 BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
BLAST Ausarbeitung zum Proseminar Vortag von Nicolás Fusseder am 24.10.02 BLAST (Basic Local Alignment Search Tool) hat seit seiner Veröffentlichung, von Altschul et al. im Jahre 1990, an großer Relevanz
Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten
 1 Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten (inkl. Anleitungen zur Recherche von Sequenzen mit GenBank und zur Analyse mit GeneDoc) In der Computer-basierten Version
1 Informationsmaterial Resistenz gegen HIV Recherche und Analyse molekularer Daten (inkl. Anleitungen zur Recherche von Sequenzen mit GenBank und zur Analyse mit GeneDoc) In der Computer-basierten Version
Vorlesungsplan. Von Naïve Bayes zu Bayesischen Netzwerk- Klassifikatoren. Naïve Bayes. Bayesische Netzwerke
 Vorlesungsplan 17.10. Einleitung 24.10. Ein- und Ausgabe 31.10. Reformationstag, Einfache Regeln 7.11. Naïve Bayes, Entscheidungsbäume 14.11. Entscheidungsregeln, Assoziationsregeln 21.11. Lineare Modelle,
Vorlesungsplan 17.10. Einleitung 24.10. Ein- und Ausgabe 31.10. Reformationstag, Einfache Regeln 7.11. Naïve Bayes, Entscheidungsbäume 14.11. Entscheidungsregeln, Assoziationsregeln 21.11. Lineare Modelle,
Verwandtschaftsbestimmung mit molekularen Daten
 Verwandtschaftsbestimmung mit molekularen Daten DITTMAR GRAF Online-Ergänzung MNU 67/5 (15.7.2014) Seiten 1 6, ISSN 0025-5866, Verlag Klaus Seeberger, Neuss 1 DITTMAR GRAF Verwandtschaftsbestimmung mit
Verwandtschaftsbestimmung mit molekularen Daten DITTMAR GRAF Online-Ergänzung MNU 67/5 (15.7.2014) Seiten 1 6, ISSN 0025-5866, Verlag Klaus Seeberger, Neuss 1 DITTMAR GRAF Verwandtschaftsbestimmung mit
Vorlesung Diskrete Strukturen Graphen: Wieviele Bäume?
 Vorlesung Diskrete Strukturen Graphen: Wieviele Bäume? Bernhard Ganter Institut für Algebra TU Dresden D-01062 Dresden bernhard.ganter@tu-dresden.de WS 2013/14 Isomorphie Zwei Graphen (V 1, E 1 ) und (V
Vorlesung Diskrete Strukturen Graphen: Wieviele Bäume? Bernhard Ganter Institut für Algebra TU Dresden D-01062 Dresden bernhard.ganter@tu-dresden.de WS 2013/14 Isomorphie Zwei Graphen (V 1, E 1 ) und (V
Statistik IV für Studenten mit dem Nebenfach Statistik Lösungen zu Blatt 9 Gerhard Tutz, Jan Ulbricht SS 07
 Statistik IV für Studenten mit dem Nebenfach Statistik Lösungen zu Blatt 9 Gerhard Tutz, Jan Ulbricht SS 07 Ziel der Clusteranalyse: Bilde Gruppen (cluster) aus einer Menge multivariater Datenobjekte (stat
Statistik IV für Studenten mit dem Nebenfach Statistik Lösungen zu Blatt 9 Gerhard Tutz, Jan Ulbricht SS 07 Ziel der Clusteranalyse: Bilde Gruppen (cluster) aus einer Menge multivariater Datenobjekte (stat
Statistische Verfahren:
 Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Statistische Verfahren: Hidden-Markov-Modelle für Multiples Alignment Stochastic Context-Free Grammars (SCFGs) für RNA-Multiples Alignment Übersicht 1 1. Hidden-Markov-Models (HMM) für Multiples Alignment
Probe-Klausur zur Vorlesung Multilinguale Mensch-Maschine Kommunikation 2013
 Probe-Klausur zur Vorlesung Multilinguale Mensch-Maschine Kommunikation 2013 Klausurnummer Name: Vorname: Matr.Nummer: Bachelor: Master: Aufgabe 1 2 3 4 5 6 7 8 max. Punkte 10 5 6 7 5 10 9 8 tats. Punkte
Probe-Klausur zur Vorlesung Multilinguale Mensch-Maschine Kommunikation 2013 Klausurnummer Name: Vorname: Matr.Nummer: Bachelor: Master: Aufgabe 1 2 3 4 5 6 7 8 max. Punkte 10 5 6 7 5 10 9 8 tats. Punkte
Graphen: Einführung. Vorlesung Mathematische Strukturen. Sommersemester 2011
 Graphen: Einführung Vorlesung Mathematische Strukturen Zum Ende der Vorlesung beschäftigen wir uns mit Graphen. Graphen sind netzartige Strukturen, bestehend aus Knoten und Kanten. Sommersemester 20 Prof.
Graphen: Einführung Vorlesung Mathematische Strukturen Zum Ende der Vorlesung beschäftigen wir uns mit Graphen. Graphen sind netzartige Strukturen, bestehend aus Knoten und Kanten. Sommersemester 20 Prof.
Mathematik 1, Teil B. Inhalt:
 FH Emden-Leer Fachb. Technik, Abt. Elektrotechnik u. Informatik Prof. Dr. J. Wiebe www.et-inf.fho-emden.de/~wiebe Mathematik 1, Teil B Inhalt: 1.) Grundbegriffe der Mengenlehre 2.) Matrizen, Determinanten
FH Emden-Leer Fachb. Technik, Abt. Elektrotechnik u. Informatik Prof. Dr. J. Wiebe www.et-inf.fho-emden.de/~wiebe Mathematik 1, Teil B Inhalt: 1.) Grundbegriffe der Mengenlehre 2.) Matrizen, Determinanten
Matrizen, Determinanten, lineare Gleichungssysteme
 Matrizen, Determinanten, lineare Gleichungssysteme 1 Matrizen Definition 1. Eine Matrix A vom Typ m n (oder eine m n Matrix, A R m n oder A C m n ) ist ein rechteckiges Zahlenschema mit m Zeilen und n
Matrizen, Determinanten, lineare Gleichungssysteme 1 Matrizen Definition 1. Eine Matrix A vom Typ m n (oder eine m n Matrix, A R m n oder A C m n ) ist ein rechteckiges Zahlenschema mit m Zeilen und n
Anmerkungen zur Übergangsprüfung
 DM11 Slide 1 Anmerkungen zur Übergangsprüfung Aufgabeneingrenzung Aufgaben des folgenden Typs werden wegen ihres Schwierigkeitsgrads oder wegen eines ungeeigneten fachlichen Schwerpunkts in der Übergangsprüfung
DM11 Slide 1 Anmerkungen zur Übergangsprüfung Aufgabeneingrenzung Aufgaben des folgenden Typs werden wegen ihres Schwierigkeitsgrads oder wegen eines ungeeigneten fachlichen Schwerpunkts in der Übergangsprüfung
Morphologische Bildverarbeitung II
 FAKULTÄT FÜR MATHEMATIK UNIVERSITÄT ULM ABT. STOCHASTIK ABT. ANGEWANDTE INFORMATIONSVERARBEITUNG Seminar Simulation und Bildanalyse mit Java Morphologische Bildverarbeitung II BETREUER: JOHANNES MAYER
FAKULTÄT FÜR MATHEMATIK UNIVERSITÄT ULM ABT. STOCHASTIK ABT. ANGEWANDTE INFORMATIONSVERARBEITUNG Seminar Simulation und Bildanalyse mit Java Morphologische Bildverarbeitung II BETREUER: JOHANNES MAYER
Die Harmonische Reihe
 Die Harmonische Reihe Wie stellt sich Determinismus in der Mathematik dar? Wie stellt man Daten dar? Wie findet man das Resultat von unendlich vielen Schritten? Mehrere Wege können zu demselben Ziel führen
Die Harmonische Reihe Wie stellt sich Determinismus in der Mathematik dar? Wie stellt man Daten dar? Wie findet man das Resultat von unendlich vielen Schritten? Mehrere Wege können zu demselben Ziel führen
9.2 Invertierbare Matrizen
 34 9.2 Invertierbare Matrizen Die Division ist als Umkehroperation der Multiplikation definiert. Das heisst, für reelle Zahlen a 0 und b gilt b = a genau dann, wenn a b =. Übertragen wir dies von den reellen
34 9.2 Invertierbare Matrizen Die Division ist als Umkehroperation der Multiplikation definiert. Das heisst, für reelle Zahlen a 0 und b gilt b = a genau dann, wenn a b =. Übertragen wir dies von den reellen
Berechnungen in Access Teil I
 in Access Teil I Viele Daten müssen in eine Datenbank nicht eingetragen werden, weil sie sich aus anderen Daten berechnen lassen. Zum Beispiel lässt sich die Mehrwertsteuer oder der Bruttopreis in einer
in Access Teil I Viele Daten müssen in eine Datenbank nicht eingetragen werden, weil sie sich aus anderen Daten berechnen lassen. Zum Beispiel lässt sich die Mehrwertsteuer oder der Bruttopreis in einer
Algorithmische Methoden zur Netzwerkanalyse
 Algorithmische Methoden zur Netzwerkanalyse Juniorprof. Dr. Henning Meyerhenke Institut für Theoretische Informatik 1 KIT Henning Universität desmeyerhenke, Landes Baden-Württemberg Institutund für Theoretische
Algorithmische Methoden zur Netzwerkanalyse Juniorprof. Dr. Henning Meyerhenke Institut für Theoretische Informatik 1 KIT Henning Universität desmeyerhenke, Landes Baden-Württemberg Institutund für Theoretische
Randomisierte Algorithmen
 Randomisierte Algorithmen Randomisierte Algorithmen 5. Zwei spieltheoretische Aspekte Thomas Worsch Fakultät für Informatik Karlsruher Institut für Technologie Wintersemester 2015/2016 1 / 36 Überblick
Randomisierte Algorithmen Randomisierte Algorithmen 5. Zwei spieltheoretische Aspekte Thomas Worsch Fakultät für Informatik Karlsruher Institut für Technologie Wintersemester 2015/2016 1 / 36 Überblick
16. All Pairs Shortest Path (ASPS)
") . All Pairs Shortest Path (ASPS) All Pairs Shortest Path (APSP): Eingabe: Gewichteter Graph G=(V,E) Ausgabe: Für jedes Paar von Knoten u,v V die Distanz von u nach v sowie einen kürzesten Weg a b c d e
. All Pairs Shortest Path (ASPS) All Pairs Shortest Path (APSP): Eingabe: Gewichteter Graph G=(V,E) Ausgabe: Für jedes Paar von Knoten u,v V die Distanz von u nach v sowie einen kürzesten Weg a b c d e
Box. Biologie. Das Nervensystem Zellbiologische Grundlagen, Erregungsbildung und Erregungsweiterleitung
 Box Biologie Schülerarbeitsbuch 2. Halbjahr der Qualifikationsphase Niedersachsen Evolution Ursachen der Evolution Evolutionstheorien Evolutive Entwicklungen Neurobiologie Das Nervensystem Zellbiologische
Box Biologie Schülerarbeitsbuch 2. Halbjahr der Qualifikationsphase Niedersachsen Evolution Ursachen der Evolution Evolutionstheorien Evolutive Entwicklungen Neurobiologie Das Nervensystem Zellbiologische
BLAST. Datenbanksuche mit BLAST. Genomische Datenanalyse 10. Kapitel
 Datenbanksuche mit BLAST BLAST Genomische Datenanalyse 10. Kapitel http://www.ncbi.nlm.nih.gov/blast/ Statistische Fragen Datenbanksuche Query Kann die globale Sequenzähnlichkeit eine Zufallsfluktuation
Datenbanksuche mit BLAST BLAST Genomische Datenanalyse 10. Kapitel http://www.ncbi.nlm.nih.gov/blast/ Statistische Fragen Datenbanksuche Query Kann die globale Sequenzähnlichkeit eine Zufallsfluktuation
Graphentheorie 1. Diskrete Strukturen. Sommersemester Uta Priss ZeLL, Ostfalia. Hausaufgaben Graph-Äquivalenz SetlX
 Graphentheorie 1 Diskrete Strukturen Uta Priss ZeLL, Ostfalia Sommersemester 2016 Diskrete Strukturen Graphentheorie 1 Slide 1/19 Agenda Hausaufgaben Graph-Äquivalenz SetlX Diskrete Strukturen Graphentheorie
Graphentheorie 1 Diskrete Strukturen Uta Priss ZeLL, Ostfalia Sommersemester 2016 Diskrete Strukturen Graphentheorie 1 Slide 1/19 Agenda Hausaufgaben Graph-Äquivalenz SetlX Diskrete Strukturen Graphentheorie
Maschinelles Lernen in der Bioinformatik
 Maschinelles Lernen in der Bioinformatik Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) VL 2 HMM und (S)CFG Jana Hertel Professur für Bioinformatik Institut für Informatik
Maschinelles Lernen in der Bioinformatik Spezialvorlesung Modul 10-202-2206 (Fortgeschrittene Methoden in der Bioinformatik) VL 2 HMM und (S)CFG Jana Hertel Professur für Bioinformatik Institut für Informatik
Berechnung phylogenetischer Bäume mit Distanzmaßen
 Berechnung phylogenetischer Bäume mit Distanzmaßen Seminar: Verwandtschaft und Abstammung in Zeichenketten WS 2004/2005 Stephan Klinger Benjamin Großmann Gliederung Einleitung...3 Geschichte der Evolutionsforschung...3
Berechnung phylogenetischer Bäume mit Distanzmaßen Seminar: Verwandtschaft und Abstammung in Zeichenketten WS 2004/2005 Stephan Klinger Benjamin Großmann Gliederung Einleitung...3 Geschichte der Evolutionsforschung...3
Optimieren unter Nebenbedingungen
 Optimieren unter Nebenbedingungen Hier sucht man die lokalen Extrema einer Funktion f(x 1,, x n ) unter der Nebenbedingung dass g(x 1,, x n ) = 0 gilt Die Funktion f heißt Zielfunktion Beispiel: Gesucht
Optimieren unter Nebenbedingungen Hier sucht man die lokalen Extrema einer Funktion f(x 1,, x n ) unter der Nebenbedingung dass g(x 1,, x n ) = 0 gilt Die Funktion f heißt Zielfunktion Beispiel: Gesucht
Prüfung Lineare Algebra Sei V ein n-dimensionaler euklidischer Raum. Welche der folgenden Aussagen ist wahr?
 1. Sei V ein n-dimensionaler euklidischer Raum. Welche der folgenden Aussagen ist wahr? A. Wenn n = 3 ist, sind mindestens zwei der drei Euler-Winkel einer Drehung kleiner oder gleich π. B. Wenn n = 2
1. Sei V ein n-dimensionaler euklidischer Raum. Welche der folgenden Aussagen ist wahr? A. Wenn n = 3 ist, sind mindestens zwei der drei Euler-Winkel einer Drehung kleiner oder gleich π. B. Wenn n = 2
Zusammenhangsanalyse in Kontingenztabellen
 Zusammenhangsanalyse in Kontingenztabellen Bisher: Tabellarische / graphische Präsentation Jetzt: Maßzahlen für Stärke des Zusammenhangs zwischen X und Y. Chancen und relative Chancen Zunächst 2 2 - Kontingenztafel
Zusammenhangsanalyse in Kontingenztabellen Bisher: Tabellarische / graphische Präsentation Jetzt: Maßzahlen für Stärke des Zusammenhangs zwischen X und Y. Chancen und relative Chancen Zunächst 2 2 - Kontingenztafel
Algebra und Diskrete Mathematik, PS3. Sommersemester Prüfungsfragen
 Algebra und Diskrete Mathematik, PS3 Sommersemester 2016 Prüfungsfragen Erläutern Sie die Sätze über die Division mit Rest für ganze Zahlen und für Polynome (mit Koeffizienten in einem Körper). Wodurch
Algebra und Diskrete Mathematik, PS3 Sommersemester 2016 Prüfungsfragen Erläutern Sie die Sätze über die Division mit Rest für ganze Zahlen und für Polynome (mit Koeffizienten in einem Körper). Wodurch
Biowissenschaftlich recherchieren
 Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
Biowissenschaftlich recherchieren Uber den Einsatz von Datenbanken und anderen Ressourcen der Bioinformatik Nicola Gaedeke Birkhauser Basel Boston Berlin Inhaltsverzeichnis Vorwort xi 1 Die Informationssucheim
Mathematische und statistische Methoden I
 Prof. Dr. G. Meinhardt 6. Stock, Wallstr. 3 (Raum 06-206) Sprechstunde jederzeit nach Vereinbarung und nach der Vorlesung. Mathematische und statistische Methoden I Dr. Malte Persike persike@uni-mainz.de
Prof. Dr. G. Meinhardt 6. Stock, Wallstr. 3 (Raum 06-206) Sprechstunde jederzeit nach Vereinbarung und nach der Vorlesung. Mathematische und statistische Methoden I Dr. Malte Persike persike@uni-mainz.de
4.4. Rang und Inversion einer Matrix
 44 Rang und Inversion einer Matrix Der Rang einer Matrix ist die Dimension ihres Zeilenraumes also die Maximalzahl linear unabhängiger Zeilen Daß der Rang sich bei elementaren Zeilenumformungen nicht ändert
44 Rang und Inversion einer Matrix Der Rang einer Matrix ist die Dimension ihres Zeilenraumes also die Maximalzahl linear unabhängiger Zeilen Daß der Rang sich bei elementaren Zeilenumformungen nicht ändert
Kapiteltests zum Leitprogramm Binäre Suchbäume
 Kapiteltests zum Leitprogramm Binäre Suchbäume Björn Steffen Timur Erdag überarbeitet von Christina Class Binäre Suchbäume Kapiteltests für das ETH-Leitprogramm Adressaten und Institutionen Das Leitprogramm
Kapiteltests zum Leitprogramm Binäre Suchbäume Björn Steffen Timur Erdag überarbeitet von Christina Class Binäre Suchbäume Kapiteltests für das ETH-Leitprogramm Adressaten und Institutionen Das Leitprogramm
Handbuch ECDL 2003 Professional Modul 2: Tabellenkalkulation Kopieren, Einfügen und Verknüpfen von Daten
 Handbuch ECDL 2003 Professional Modul 2: Tabellenkalkulation Kopieren, Einfügen und Verknüpfen von Daten Dateiname: ecdl_p2_02_02_documentation.doc Speicherdatum: 08.12.2004 ECDL 2003 Professional Modul
Handbuch ECDL 2003 Professional Modul 2: Tabellenkalkulation Kopieren, Einfügen und Verknüpfen von Daten Dateiname: ecdl_p2_02_02_documentation.doc Speicherdatum: 08.12.2004 ECDL 2003 Professional Modul
Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut, keine vorgegebenen Klassen
 7. Clusteranalyse (= Häufungsanalyse; Clustering-Verfahren) wird der multivariaten Statistik zugeordnet Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut,
7. Clusteranalyse (= Häufungsanalyse; Clustering-Verfahren) wird der multivariaten Statistik zugeordnet Voraussetzung wieder: Datenraum mit Instanzen, mehrere Attribute - kein ausgezeichnetes Zielattribut,
Maximizing the Spread of Influence through a Social Network
 1 / 26 Maximizing the Spread of Influence through a Social Network 19.06.2007 / Thomas Wener TU-Darmstadt Seminar aus Data und Web Mining bei Prof. Fürnkranz 2 / 26 Gliederung Einleitung 1 Einleitung 2
1 / 26 Maximizing the Spread of Influence through a Social Network 19.06.2007 / Thomas Wener TU-Darmstadt Seminar aus Data und Web Mining bei Prof. Fürnkranz 2 / 26 Gliederung Einleitung 1 Einleitung 2
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
Bio Data Management Kapitel 5a Sequenzierung und Alignments Sommersemester 2013 Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges Inhaltsverzeichnis
6. Faktorenanalyse (FA) von Tests
 von Tests") 6. Faktorenanalyse (FA) von Tests 1 6. Faktorenanalyse (FA) von Tests 1 6.1. Grundzüge der FA nach der Haupkomponentenmethode (PCA) mit anschliessender VARIMAX-Rotation:... 2 6.2. Die Matrizen der FA...
6. Faktorenanalyse (FA) von Tests 1 6. Faktorenanalyse (FA) von Tests 1 6.1. Grundzüge der FA nach der Haupkomponentenmethode (PCA) mit anschliessender VARIMAX-Rotation:... 2 6.2. Die Matrizen der FA...
Algorithmen und Datenstrukturen 2
 Algorithmen und Datenstrukturen 2 Sommersemester 2007 4. Vorlesung Peter F. Stadler Universität Leipzig Institut für Informatik studla@bioinf.uni-leipzig.de Traversierung Durchlaufen eines Graphen, bei
Algorithmen und Datenstrukturen 2 Sommersemester 2007 4. Vorlesung Peter F. Stadler Universität Leipzig Institut für Informatik studla@bioinf.uni-leipzig.de Traversierung Durchlaufen eines Graphen, bei
Lineare Gleichungssysteme (Teschl/Teschl 11.1)
") Lineare Gleichungssysteme (Teschl/Teschl.) Ein Lineares Gleichungssystem (LGS) besteht aus m Gleichungen mit n Unbekannten x,...,x n und hat die Form a x + a 2 x 2 +... + a n x n b a 2 x + a 22 x 2 +...
Lineare Gleichungssysteme (Teschl/Teschl.) Ein Lineares Gleichungssystem (LGS) besteht aus m Gleichungen mit n Unbekannten x,...,x n und hat die Form a x + a 2 x 2 +... + a n x n b a 2 x + a 22 x 2 +...
Satz 16 (Multiplikationssatz)
") Häufig verwendet man die Definition der bedingten Wahrscheinlichkeit in der Form Damit: Pr[A B] = Pr[B A] Pr[A] = Pr[A B] Pr[B]. (1) Satz 16 (Multiplikationssatz) Seien die Ereignisse A 1,..., A n gegeben.
Häufig verwendet man die Definition der bedingten Wahrscheinlichkeit in der Form Damit: Pr[A B] = Pr[B A] Pr[A] = Pr[A B] Pr[B]. (1) Satz 16 (Multiplikationssatz) Seien die Ereignisse A 1,..., A n gegeben.
Einführung in die Kodierungstheorie
 Einführung in die Kodierungstheorie Einführung Vorgehen Beispiele Definitionen (Code, Codewort, Alphabet, Länge) Hamming-Distanz Definitionen (Äquivalenz, Coderate, ) Singleton-Schranke Lineare Codes Hamming-Gewicht
Einführung in die Kodierungstheorie Einführung Vorgehen Beispiele Definitionen (Code, Codewort, Alphabet, Länge) Hamming-Distanz Definitionen (Äquivalenz, Coderate, ) Singleton-Schranke Lineare Codes Hamming-Gewicht
A.12 Nullstellen / Gleichungen lösen
 A12 Nullstellen 1 A.12 Nullstellen / Gleichungen lösen Es gibt nur eine Hand voll Standardverfahren, nach denen man vorgehen kann, um Gleichungen zu lösen. Man sollte in der Gleichung keine Brüche haben.
A12 Nullstellen 1 A.12 Nullstellen / Gleichungen lösen Es gibt nur eine Hand voll Standardverfahren, nach denen man vorgehen kann, um Gleichungen zu lösen. Man sollte in der Gleichung keine Brüche haben.
Lösungen zu den Übungsaufgaben aus Kapitel 3
 Lösungen zu den Übungsaufgaben aus Kapitel 3 Ü3.1: a) Die Start-Buchungslimits betragen b 1 = 25, b 2 = 20 und b 3 = 10. In der folgenden Tabelle sind jeweils die Annahmen ( ) und Ablehnungen ( ) der Anfragen
Lösungen zu den Übungsaufgaben aus Kapitel 3 Ü3.1: a) Die Start-Buchungslimits betragen b 1 = 25, b 2 = 20 und b 3 = 10. In der folgenden Tabelle sind jeweils die Annahmen ( ) und Ablehnungen ( ) der Anfragen
Uninformierte Suche in Java Informierte Suchverfahren
 Uninformierte Suche in Java Informierte Suchverfahren Stephan Schwiebert WS 2009/2010 Sprachliche Informationsverarbeitung Institut für Linguistik Universität zu Köln Suchprobleme bestehen aus Zuständen
Uninformierte Suche in Java Informierte Suchverfahren Stephan Schwiebert WS 2009/2010 Sprachliche Informationsverarbeitung Institut für Linguistik Universität zu Köln Suchprobleme bestehen aus Zuständen
Literatur. Dominating Set (DS) Dominating Sets in Sensornetzen. Problem Minimum Dominating Set (MDS)
 Dominating Sets in Sensornetzen. Problem Minimum Dominating Set (MDS)") Dominating Set 59 Literatur Dominating Set Grundlagen 60 Dominating Set (DS) M. V. Marathe, H. Breu, H.B. Hunt III, S. S. Ravi, and D. J. Rosenkrantz: Simple Heuristics for Unit Disk Graphs. Networks 25,
Dominating Set 59 Literatur Dominating Set Grundlagen 60 Dominating Set (DS) M. V. Marathe, H. Breu, H.B. Hunt III, S. S. Ravi, and D. J. Rosenkrantz: Simple Heuristics for Unit Disk Graphs. Networks 25,
Bio Data Management. Kapitel 5a Sequenzierung und Alignments
 Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Bio Data Management Kapitel 5a Sequenzierung und Alignments Wintersemester 2014/15 Dr. Anika Groß Universität Leipzig, Institut für Informatik, Abteilung Datenbanken http://dbs.uni-leipzig.de Vorläufiges
Datenkompression. 1 Allgemeines. 2 Verlustlose Kompression. Holger Rauhut
 Datenkompression Holger Rauhut 1. September 2010 Skript für die Schülerwoche 2010, 8.-11.9.2010 Hausdorff Center for Mathematics, Bonn 1 Allgemeines Datenkompression hat zum Ziel, Daten in digitaler Form,
Datenkompression Holger Rauhut 1. September 2010 Skript für die Schülerwoche 2010, 8.-11.9.2010 Hausdorff Center for Mathematics, Bonn 1 Allgemeines Datenkompression hat zum Ziel, Daten in digitaler Form,
Korrelationsmatrix. Statistische Bindungen zwischen den N Zufallsgrößen werden durch die Korrelationsmatrix vollständig beschrieben:
 Korrelationsmatrix Bisher wurden nur statistische Bindungen zwischen zwei (skalaren) Zufallsgrößen betrachtet. Für den allgemeineren Fall einer Zufallsgröße mit N Dimensionen bietet sich zweckmäßiger Weise
Korrelationsmatrix Bisher wurden nur statistische Bindungen zwischen zwei (skalaren) Zufallsgrößen betrachtet. Für den allgemeineren Fall einer Zufallsgröße mit N Dimensionen bietet sich zweckmäßiger Weise
Venndiagramm, Grundmenge und leere Menge
 Venndiagramm, Grundmenge und leere Menge In späteren Kapitel wird manchmal auf die Mengenlehre Bezug genommen. Deshalb sollen hier die wichtigsten Grundlagen und Definitionen dieser Disziplin kurz zusammengefasst
Venndiagramm, Grundmenge und leere Menge In späteren Kapitel wird manchmal auf die Mengenlehre Bezug genommen. Deshalb sollen hier die wichtigsten Grundlagen und Definitionen dieser Disziplin kurz zusammengefasst
Lineare Gleichungssysteme
 Brückenkurs Mathematik TU Dresden 2015 Lineare Gleichungssysteme Schwerpunkte: Modellbildung geometrische Interpretation Lösungsmethoden Prof. Dr. F. Schuricht TU Dresden, Fachbereich Mathematik auf der
Brückenkurs Mathematik TU Dresden 2015 Lineare Gleichungssysteme Schwerpunkte: Modellbildung geometrische Interpretation Lösungsmethoden Prof. Dr. F. Schuricht TU Dresden, Fachbereich Mathematik auf der
3. Einführung in die Theorie der Methoden
 3. Einführung in die Theorie der Methoden 3.1. Morphologische Daten Die Analyse morphologischer Merkmale bildet auch heute die Grundlage phylogenetischer Untersuchungen. Morphologische Merkmale sind der
3. Einführung in die Theorie der Methoden 3.1. Morphologische Daten Die Analyse morphologischer Merkmale bildet auch heute die Grundlage phylogenetischer Untersuchungen. Morphologische Merkmale sind der
Ein Algorithmus für die
 VGG 1 Ein Algorithmus für die Visualisierung gerichteter Graphen in der Ebene (2D) Seminar Graph Drawing SS 2004 bei Prof. Bischof (Lehrstuhl für Hochleistungsrechnen) Gliederung VGG 2 Einleitung Motivation
VGG 1 Ein Algorithmus für die Visualisierung gerichteter Graphen in der Ebene (2D) Seminar Graph Drawing SS 2004 bei Prof. Bischof (Lehrstuhl für Hochleistungsrechnen) Gliederung VGG 2 Einleitung Motivation
Einführung in Quantencomputer
 Einführung in Quantencomputer Literatur M. Homeister, (jetzt FB Informatik und Medien an der Fachhochschule Brandenburg) Quantum Computing verstehen, Springer Vieweg Verlag (25) E. Rieffel und W. Polak,
Einführung in Quantencomputer Literatur M. Homeister, (jetzt FB Informatik und Medien an der Fachhochschule Brandenburg) Quantum Computing verstehen, Springer Vieweg Verlag (25) E. Rieffel und W. Polak,
Korrelation und Regression
 FB 1 W. Ludwig-Mayerhofer und 1 und FB 1 W. Ludwig-Mayerhofer und 2 Mit s- und sanalyse werden Zusammenhänge zwischen zwei metrischen Variablen analysiert. Wenn man nur einen Zusammenhang quantifizieren
FB 1 W. Ludwig-Mayerhofer und 1 und FB 1 W. Ludwig-Mayerhofer und 2 Mit s- und sanalyse werden Zusammenhänge zwischen zwei metrischen Variablen analysiert. Wenn man nur einen Zusammenhang quantifizieren
7 Lineare Gleichungssysteme
 118 7 Lineare Gleichungssysteme Lineare Gleichungssysteme treten in vielen mathematischen, aber auch naturwissenschaftlichen Problemen auf; zum Beispiel beim Lösen von Differentialgleichungen, bei Optimierungsaufgaben,
118 7 Lineare Gleichungssysteme Lineare Gleichungssysteme treten in vielen mathematischen, aber auch naturwissenschaftlichen Problemen auf; zum Beispiel beim Lösen von Differentialgleichungen, bei Optimierungsaufgaben,
Kapitel 2: Matrizen. 2.1 Matrizen 2.2 Determinanten 2.3 Inverse 2.4 Lineare Gleichungssysteme 2.5 Eigenwerte 2.6 Diagonalisierung
 Kapitel 2: Matrizen 2.1 Matrizen 2.2 Determinanten 2.3 Inverse 2.4 Lineare Gleichungssysteme 2.5 Eigenwerte 2.6 Diagonalisierung 2.1 Matrizen M = n = 3 m = 3 n = m quadratisch M ij : Eintrag von M in i-ter
Kapitel 2: Matrizen 2.1 Matrizen 2.2 Determinanten 2.3 Inverse 2.4 Lineare Gleichungssysteme 2.5 Eigenwerte 2.6 Diagonalisierung 2.1 Matrizen M = n = 3 m = 3 n = m quadratisch M ij : Eintrag von M in i-ter
Lineare Gleichungssysteme
 Christian Serpé Universität Münster 14. September 2011 Christian Serpé (Universität Münster) 14. September 2011 1 / 56 Gliederung 1 Motivation Beispiele Allgemeines Vorgehen 2 Der Vektorraum R n 3 Lineare
Christian Serpé Universität Münster 14. September 2011 Christian Serpé (Universität Münster) 14. September 2011 1 / 56 Gliederung 1 Motivation Beispiele Allgemeines Vorgehen 2 Der Vektorraum R n 3 Lineare
Bioinformatik I (Einführung)
") Kay Diederichs, Sommersemester 2015 Bioinformatik I (Einführung) Algorithmen Sequenzen Strukturen PDFs unter http://strucbio.biologie.unikonstanz.de/~dikay/bioinformatik/ Klausur: Fr 17.7. 10:00-11:00
Kay Diederichs, Sommersemester 2015 Bioinformatik I (Einführung) Algorithmen Sequenzen Strukturen PDFs unter http://strucbio.biologie.unikonstanz.de/~dikay/bioinformatik/ Klausur: Fr 17.7. 10:00-11:00
Phishingerkennung mittels visuellem Ähnlichkeitsvergleich. Felix Hill Ruhr-Universität Bochum felix.hill@rub.de
 Phishingerkennung mittels visuellem Ähnlichkeitsvergleich Felix Hill Ruhr-Universität Bochum felix.hill@rub.de 1 ÜBERSICHT Entwicklung im Bereich Phishing Ansatz Bilderkennung Evaluation G DATA EINFACH
Phishingerkennung mittels visuellem Ähnlichkeitsvergleich Felix Hill Ruhr-Universität Bochum felix.hill@rub.de 1 ÜBERSICHT Entwicklung im Bereich Phishing Ansatz Bilderkennung Evaluation G DATA EINFACH
MC-Serie 11: Eigenwerte
 D-ERDW, D-HEST, D-USYS Mathematik I HS 14 Dr. Ana Cannas MC-Serie 11: Eigenwerte Einsendeschluss: 12. Dezember 2014 Bei allen Aufgaben ist genau eine Antwort richtig. Lösens des Tests eine Formelsammlung
D-ERDW, D-HEST, D-USYS Mathematik I HS 14 Dr. Ana Cannas MC-Serie 11: Eigenwerte Einsendeschluss: 12. Dezember 2014 Bei allen Aufgaben ist genau eine Antwort richtig. Lösens des Tests eine Formelsammlung
3. Entscheidungsbäume. Verfahren zum Begriffslernen (Klassifikation) Beispiel: weiteres Beispiel: (aus Böhm 2003) (aus Morik 2002)
 Beispiel: weiteres Beispiel: (aus Böhm 2003) (aus Morik 2002)") 3. Entscheidungsbäume Verfahren zum Begriffslernen (Klassifikation) Beispiel: weiteres Beispiel: (aus Böhm 2003) (aus Morik 2002) (aus Wilhelm 2001) Beispiel: (aus Böhm 2003) Wann sind Entscheidungsbäume
3. Entscheidungsbäume Verfahren zum Begriffslernen (Klassifikation) Beispiel: weiteres Beispiel: (aus Böhm 2003) (aus Morik 2002) (aus Wilhelm 2001) Beispiel: (aus Böhm 2003) Wann sind Entscheidungsbäume
Bioinformatik. Dynamische Programmierung. Ulf Leser Wissensmanagement in der. Bioinformatik
 Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Bioinformatik Dynamische Programmierung Ulf Leser Wissensmanagement in der Bioinformatik Motivation BLAST / FASTA und Verwandte sind *die* Bioinformatik Anwendung Teilweise synonym für Bioinformatik rundlegende
Einführung in die Vektor- und Matrizenrechnung. Matrizen
 Einführung in die Vektor- und Matrizenrechnung Matrizen Definition einer Matrix Unter einer (reellen) m x n Matrix A versteht man ein rechteckiges Schema aus reellen Zahlen, die wie folgt angeordnet sind:
Einführung in die Vektor- und Matrizenrechnung Matrizen Definition einer Matrix Unter einer (reellen) m x n Matrix A versteht man ein rechteckiges Schema aus reellen Zahlen, die wie folgt angeordnet sind:
Prof. Dr. Christoph Kleinn Institut für Waldinventur und Waldwachstum Arbeitsbereich Waldinventur und Fernerkundung
 Systematische Stichprobe Rel. große Gruppe von Stichprobenverfahren. Allgemeines Merkmal: es existiert ein festes, systematisches Muster bei der Auswahl. Wie passt das zur allgemeinen Forderung nach Randomisierung
Systematische Stichprobe Rel. große Gruppe von Stichprobenverfahren. Allgemeines Merkmal: es existiert ein festes, systematisches Muster bei der Auswahl. Wie passt das zur allgemeinen Forderung nach Randomisierung
5.1 Determinanten der Ordnung 2 und 3. a 11 a 12 a 21 a 22. det(a) =a 11 a 22 a 12 a 21. a 11 a 21
 =a 11 a 22 a 12 a 21. a 11 a 21") 5. Determinanten 5.1 Determinanten der Ordnung 2 und 3 Als Determinante der zweireihigen Matrix A = a 11 a 12 bezeichnet man die Zahl =a 11 a 22 a 12 a 21. Man verwendet auch die Bezeichnung = A = a 11
5. Determinanten 5.1 Determinanten der Ordnung 2 und 3 Als Determinante der zweireihigen Matrix A = a 11 a 12 bezeichnet man die Zahl =a 11 a 22 a 12 a 21. Man verwendet auch die Bezeichnung = A = a 11