Induktion eines alternativen Telomererhaltungsmechanismus in humanen Tumoren durch genetische Inhibition der Telomerase
|
|
|
- Horst Knopp
- vor 6 Jahren
- Abrufe
Transkript


1 Institut für Molekulare Medizin und Zellforschung Freiburg im Breisgau Induktion eines alternativen Telomererhaltungsmechanismus in humanen Tumoren durch genetische Inhibition der Telomerase Inaugural-Dissertation Zur Erlangung der Doktorwürde der Fakultät für Biologie der Albert-Ludwigs-Universität Freiburg im Breisgau vorgelegt von Angela Queisser aus Ludwigsburg Dezember 2010
2 Die vorliegende Arbeit wurde im Zeitraum von Januar 2006 bis Dezember 2010 am Institut für Molekulare Medizin und Zellforschung in Freiburg im Breisgau unter Betreuung von Prof. Dr. Oliver G. Opitz angefertigt. Dekan der Fakultät: Promotionsvorsitzender: Prof. Dr. Gunther Neuhaus Prof. Dr. Samuel Rossel Betreuer: Prof. Dr. Oliver G. Opitz Referent: Prof. Dr. Christoph Peters Koreferent: PD Dr. Ekkehard Schulze 3. Prüfer: Prof. Dr. Oliver G. Opitz Tag der mündlichen Prüfung:
3
4 Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis... I 1 Einleitung Aufbau und Funktion von Telomeren Telomererhaltungsmechanismen Telomerase Telomeraseaktivierung führt zur Immortalisierung von normalen Zellen Der alternative Mechanismus zur Verlängerung der Telomere Charakteristika von ALT-positiven Zellen ALT-Mechanismus und Telomerrekombination Post-replikativer Telomeraustausch zwischen Chromosomen und Schwester-chromatiden Kopieren von Telomeren über homologe Rekombination Telomererhaltungsproteine in ALT-Zellen Bedeutung der Telomererhaltung in der Tumorgenese Das Plattenepithelkarzinom des Ösophagus als Modell zur Untersuchung der Telomerbiologie Das Adenokarzinom des Ösophagus Zielsetzung Material und Methoden Material Chemikalien Sonstiges Sonstige Reagenzien Einmalartikel: Plastik und Glaswaren Reaktionskits Geräte Mikroskope Zentrifugen Elektrophoresekammern Programme Sonstiges Enzyme Zelllinien Plasmide Lentivirale Plasmide: Telomeraseinhibitoren I
5 Inhaltsverzeichnis Antikörper Telomer-Sonden Oligonukleotide Puffer und Lösungen Zellkultur Zellkulturmedien Einfriermedium Molekularbiologie Biochemie Indirekte Immunfluoreszenz Fluoreszenz in situ Hybridisierung (FISH) Methoden Zellbiologische Methoden Kultivieren von Zellen Zellen einfrieren Zellen auftauen Wachstumskurven erstellen Herstellung von Viruspartikeln Lentivirale Transduktion (Spin Transfection) Molekularbiologische Methoden Telomere Restriction Amplification Protocol (TRAP) DNA Extraktion Messung von DNA Telomere Restriction Fragments (TRF) Methylierungsspezifische PCR (MSP) Biochemische Methoden Proteinextraktion mit CHAPS-Puffer Proteinbestimmung nach Bradford SDS-Polyacrylamidgelelektrophorese (SDS-PAGE) Transfer von Proteinen auf PVDF-Membranen Immunblotanalyse und Detektion durch verstärkte Chemilumineszenz (ECL) Indirekte Immunfluoreszenz ALT-assoziierte PML-Bodies (APBs) Fluoreszenz in situ Hybridisierung (FISH) Quantitative Fluoreszenz in situ Hybridisierung (Q-FISH) Chromosomen Orientierungs- Fluoreszenz in situ Hybridisierung (CO-FISH) Fluoreszenz-aktivierendes Zellsortieren (FACS) II
6 Inhaltsverzeichnis 3 Ergebnisse Charakterisierung der verschiedenen Zelllinien Charakterisierung des Barrettzell-Modells Charakterisierung des zellulären Plattenepithelkarzinogenese-Modells Charakterisierung der Telomeraseaktivität und der Telomerlängen in der genetisch definierten ösophagealen Plattenepithelkarzinomzelllinie, der genetisch definierten immortalen Barrettzelllinie und der genetisch nicht definierten Adenokarzinomzelllinie Genetische Inhibition der Telomerase führt zur Induktion eines klassischen ALT- Mechanismus in genetisch definierten Zelllinien, aber nicht in der genetisch nicht definierten Adenokarzinomzellen Genetische Inhibition der Telomerase führt zur Reduktion der Telomeraseaktivität und zum Erhalt von Telomeren in genetisch definierten Zellen Untersuchungen der Telomeraseaktivität und Telomerlängen in genetisch definierten Plattenepithelkarzinomzellen Untersuchung der Telomeraseaktivität und Telomerlängen in den genetisch definierten Barrettzellen Untersuchungen der Telomeraseaktivität und Telomerlängen in den genetisch nicht definierten Adenokarzinomzellen Zellmorphologische Charakterisierung der Telomerase-inhibierten Zellen Wachstumsverhalten der Telomerase-inhibierten Zellen Telomerlängenbestimmung auf Einzelzellbasis mittels Q-FISH-Analyse Nachweis von Rekombinationsereignissen an Telomeren zwischen Tochterchromatiden anhand von Chromosomen-Orientierungs-Fluoreszenz in situ Hybridisierung ALT-assoziierte PML-Bodies (APBs) in genetisch definierten immortalisierten und maligne transformierten Zellen sowie in nicht definierten Tumorzellen Diskussion Charakterisierung der verschiedenen Zelllinien Inhibierung der Telomerase Einfluss von Telomeraseinhibitoren auf die Telomererhaltung Telomeraseinhibierung führt zur Reduktion der Telomeraseaktivität in allen Zelllinien Telomeraseinhibition führt zu ALT-typischen Telomerlängen in den genetisch definierten Zellen Telomeraseinhibierung führt zur Abnahme der Telomerlängen in den genetisch nicht definierten Adenokarzinomzellen III
7 Inhaltsverzeichnis Telomeraseinhibierung führt weder zum Wachstumsstopp noch zur Apoptose in genetisch definierten immortalisierten Zellen oder Tumorzellen sowie in genetisch nicht definierten Tumorzellen Q-FISH-Analysen bestätigen heterogene Telomerlängen in allen untersuchten Telomerase-inhibierten Zelllinien Eine Telomeraseinhibierung führt nicht zu einer Erhöhung des Schwesterchromatidaustausches in allen Zelllinien Erhöhtes Auftreten von APBs in Telomerase-inhibierten genetisch definierten Zellen Telomeraseinhibierte Zellen können auf einen ALT-Mechanismus wechseln Unterschiedliche ALT-Mechanismen in unterschiedlichen Tumorentitäten und Tumorprogressionsstufen Telomeraseinhibierung führt zur Induktion von ALT: Auswirkungen auf die Behandlung von Tumorzellen mit Telomeraseinhibitoren Perspektiven Zusammenfassung und Ausblick Literaturverzeichnis Abkürzungsverzeichnis Publikationsliste Publikationen Konferenzen (Auswahl) IV
8 1 Einleitung 1 Einleitung 1.1 Aufbau und Funktion von Telomeren Lineare Chromosomen kommen in allen eukaryotischen Zellen vor und an deren Enden befindet sich eine spezialisierte Struktur, die 1938 erstmals von Herman Muller in Drosophila melanogaster beschrieben und als Telomer bezeichnet wurde (Muller, 1938). Unabhängig davon beschrieb ebenso Barbara McClintock diese Struktur in Zea mays (McClintock, 1941). Telomere bestehen aus kurzen repetitiven G-reichen Basensequenzen, die sich je nach Spezies stark unterscheiden können, jedoch keine genetische Information tragen (Klapper et al., 2001). Bei Vertebraten bestehen diese aus (TTAGGG) n -Hexamer-Wiederholungen und haben in normalen humanen somatischen Zellen eine Länge von 4-12 kbp (Moyzis et al., 1988). Am Ende der Telomere befindet sich ein G-reicher 3 -Überhang von Nukleotiden (Makarov et al., 1997; Sfeir et al., 2005), der wiederum durch Umschlagen in die proximale Telomer-Doppelstrang-DNA invadiert und somit eine Sekundärstruktur, die so genannte T-Loop (Telomer Loop), ausbildet (Griffith et al., 1999). Die verlagerte Doppelstrang-DNA, die durch Eindringen der G-reichen Einzelstrang-DNA zustande kommt formt hingegen eine so genannte D-Loop (Abbildung 1.1 B) (de Lange, 2005; Griffith et al., 1999). Wie in Abbildung 1.1 dargestellt, tragen sechs Proteine, die Proteine des so genannten Shelterin-Komplexes oder des Telosoms, zur Stabilisierung der T-Loop-Struktur bei (de Lange, 2005). Drei dieser Proteine interagieren, direkt mit der Telomer-DNA: TRF1 und 2 (Telomeric Repeat Binding Factors 1 und 2) binden als Homodimere nur spezifisch an die Telomer-DNA während Pot1 (Protection of telomeres 1), nur an den Einzelstrang 3 - Telomerüberhang binden kann. Die übrigen drei Proteinen des Shelterin-Komplexes Tin2 (TRF2- und TRF1-interacting nuclear protein 2), Rap1 (Repressor/activator protein 1) und Tpp1, interagieren nicht direkt mit der Telomer-DNA. TRF1 und TRF2 wiederum rekrutieren die anderen vier Proteine des Shelterin-Komplexes zu den Telomeren (Palm und de Lange, 2008). Die Proteine des Shelterin-Komplexes und weitere Proteine sind essentiell an der Telomerlängenregulation, Integrität und Funktion beteiligt (Blasco und Hahn, 2003). Schlüsselfunktionen dieser T-Loop-Struktur sind der Schutz vor End-zu-End-Fusionen, Degradationen und Translokationen (Blackburn, 1991). Sie versteckt sonst freiliegende Chromosomenenden und schützt so die Telomere vor der DNA-Doppelstrangbruch- Maschinerie (Ferreira et al., 2004; Griffith et al., 1999), wie z.b. der Aktivierung von Signalkaskaden wie ATM, p53 und p21, die letztlich zum Zellzyklusarrest oder zur Apoptose führen (de Lange, 2002; Karlseder et al., 2002). Man spricht hier von dem so genannten Capping der Telomere (de Lange, 2001; Munoz-Jordan et al., 2001). 1
9 1 Einleitung Außerdem nehmen Telomere eine wichtige Rolle während der Meiose und Mitose ein. Dort dient die Kernmatrix als Anheftungsstelle für Telomere, um so eine spezifische Position der Chromosomenpaare und die Bewegung der Chromosomen während der Mitose und Meiose zu gewährleisten (Klapper et al., 2001; Zickler und Kleckner, 1998). A Pot1 Tpp1 Pot1 Pot1 Tin2 Tpp1 Tpp1 TRF1 TRF2 Rap1 Subtelomer Telomer B T-Loop D-Loop Abb. 1.1: Aufbau der Telomere (A) Interaktion der Shelterin-Proteine mit der linearen Telomer-DNA. TRF1 und TRF2 interagieren direkt mit der Telomer-Doppelstrang-DNA und rekrutieren die Proteine Tpp1, Tin2, Rap1 und Pot1. An den 3 - Überhang bindet Pot1. (B) Vereinfachte Darstellung der T-Loop-Struktur. Der 3 -Überhang invadiert in den Telomer-Doppelstrang und bildet so die Struktur der T-Loop aus. Die Proteine des Shelterin- Komplexes und weitere Proteine (nicht dargestellt) stabilisieren diese Struktur. Durch das Eindringen der Einzelstrang-DNA in den Doppelstrang, wird dieser verdrängt und bildet eine D-Loop-Struktur aus. Abbildung verändert nach Gonzales-Suarez (Gonzalez-Suarez und Gonzalo, 2008). Unter normalen Bedingungen ist jedoch eine der bekanntesten und wichtigsten Funktion der Telomere das Vorbeugen des Verlusts von Erbinformationen bei der DNA-Replikation und Zellteilung, denn in den meisten humanen somatischen Zellen kommt es zu einem Verlust von ungefähr bp pro Zellteilung (Muntoni und Reddel, 2005). Grund hierfür ist das so genannte End-Replikationsproblem. Es kommt dadurch zustande, dass die Verlängerung des Tochter-DNA-Strangs durch die DNA-Polymerase nur in Richtung 5-3 am Vorwärtsstrang erfolgen kann. Der Rückwärtsstrang kann nur entgegengesetzt der Replikation diskontinuierlich in Stücken gebildet werden (Ogawa und Okazaki, 1980). Okazaki konnte 2
10 1 Einleitung 1965 als erster zeigen, dass ein Teil der frisch synthetisierten DNA in Form von kurzen Fragmenten (Okazaki-Fragmenten) vorliegt, die beim Menschen aus 200 Nukleotiden bestehen. Die Fragmente werden mit Hilfe von RNA-Primern gebildet, die dann schließlich die DNA-Synthese initiieren. Diese werden dann durch DNA-Nukleotide mit Hilfe der DNA- Polymerase ersetzt, die so Lücken aufgefüllt und schließlich mittels Ligase zum vollständigen DNA-Strang miteinander verbunden (Olovnikov, 1996). Das Problem ist jedoch, dass die Primer nicht am direkten 5 -Ende, ansetzten können, was nach jeder Replikation, wie in Abbildung 1.2 dargestellt, zur Verkürzung des neusynthetisierten Tochter-Strangs führt (Klapper et al., 2001; Olovnikov, 1996) DNA-Polymerase 5 3 Okazaki-Fragment/ RNA-Primer Vorwärtsstrang 3 5 Rückwärtsstrang Abb. 1.2 Schematische Darstellung des End-Replikationsproblems Doppelstrang-DNA (oben) wird mit Hilfe der Replikationsgabel über das komplette Chromosom repliziert (Mitte). RNA-Primer dienen als Startpunkt für die DNA-Polymerase und werden anschließend, außer an den 5 -Enden, überall durch DNA ersetzt (unten). Folglich wird der neusynthetisierte Strang nach jeder Zellteilung um bp verkürzt. Abbildung verändert nach Klapper (2001) (Klapper et al., 2001) Die Telomerlänge stellt somit eine Limitierung der möglichen Anzahl an Zellteilungen innerhalb einer Zelle dar. Wenn die Telomere eine kritisch kurze Länge erreichen, verlassen die Zellen den Zellzyklus und es wird ein Ruhezustand, die so genannte replikative Seneszenz eingeleitet. Hierdurch wird z.b. nicht nur eine chromosomale Instabilität verhindert (Hanahan und Weinberg, 2000), sondern auch der programmierte Zelltod, die Apoptose (Karlseder et al., 1999). Dieser replikationsabhängige Verlust der Telomere wurde von Harley 3
11 1 Einleitung als mitotische Uhr beschrieben (Harley et al., 1990). Hierbei ist nicht nur der reine Verlust von Telomersequenzen sondern auch das Auflösen der T-Loop-Struktur von Bedeutung (Blackburn, 2001). So konnte gezeigt werden, dass eine Überexpression von dominant negativem TRF2, welches essentiell zur Bildung der T-Loop-Struktur beiträgt (Griffith et al., 1999; Stansel et al., 2001), zur Aktivierung des p16/rb- oder ATM/p53-Signalweg führt und dadurch eine zelluläre Seneszenz oder Apoptose ausgelöst wird (Karlseder et al., 1999; Smogorzewska und de Lange, 2002). Des Weiteren führte die Inhibition von TRF2 durch Öffnen der T-Loop-Struktur zu End-zu-End-Fusionen und chromosomaler Instabilität (Karlseder et al., 2002). Ein weiterer Kontrollpunkt, nach der replikativen Seneszenz, der bei kritischen Telomeren erreicht wird, ist die so genannte Krise (Abb.1.3). Hierbei sterben fast alle Zellen, bedingt durch extrem kurze Telomere, an chromosomalen Aberrationen. Zellen können diesen Status der Krise nur überstehen, indem sie immortal werden (Klapper et al., 2001). Um diese Immortalität erreichen zu können, müssen die Zellen einen Telomererhaltungsmechanismus wie das Enzym Telomerase oder den ALT-Mechanismus (Alternative Lengthening of Telomeres) aktivieren (Abb. 1.3). Telomerlänge 2-4 kb 5-8 kb 15 kb Seneszenz Apoptose immortale Zelle +Telomerase +ALT Seneszenz Krise Zellteilungen Abb. 1.3 Die Telomer-Hypothese Die Telomere werden in somatischen Zellen auf Grund des End-Replikationsproblems mit jeder Zellteilung kürzer. Bei einer bestimmten Länge treten sie aus dem Zellzyklus aus und gehen in ein seneszentes Stadium über. Einige Zellen können diesen Kontrollpunkt übergehen, wenn bei ihnen der p53- und RB-Signalweg inaktiviert ist. Trotzdem sterben die meisten Zellen, bevor sie das so genannte Stadium der Krise erreichen. Um immortal zu werden, muss diese überschritten werden und dafür muss die Zelle einen Telomererhaltungsmechanismus aktivieren. Abbildung verändert nach Klapper (2001) (Klapper et al., 2001). 4
12 1 Einleitung 1.2 Telomererhaltungsmechanismen Telomerase Wie in Abbildung 1.3 dargestellt, benötigen immortale Zellen wie Tumorzellen einen Telomererhaltungsmechanismus, um die Seneszenz bzw. Apoptose zu umgehen. Der bekannteste Telomererhaltungsmechanismus ist die Aktivierung des Enzyms Telomerase wurde von Carol Greider und Elisabeth Blackburn erstmals ein Enzym beschrieben, das sie zunächst als telomere terminal transferase und schließlich dann als Telomerase bezeichneten. Sie konnten zeigen, dass sich im Tetrahymena-Kernextrakt eine Aktivität befand, die in der Lage war ein synthetisches Telomer-Oligonukleotid (TTGGGG) 4 zu verlängern (Greider und Blackburn, 1985). Behandlung mit RNase inaktivierte die Telomerase, so dass man davon ausging, dass ein RNA-Molekül als Matrize für das Anheften von Nukleotiden verantwortlich ist (Greider und Blackburn, 1987). Heute weiß man, dass die humane Telomerase, die ein Ribonukleoproteinkomplex darstellt, aus einer katalytischen Untereinheit, der humanen Telomerase reversen Transkriptase (htert), aus einem 451 Nukleotid langen RNA-Molekül (hter), das die Matrize für die neuzubildende Telomersequenz enthält, aus dem Protein Dyskerin, das in die Reifung des Ribonukleoproteins verwickelt ist, und weiteren Proteinen besteht (Feng et al., 1995; Mitchell et al., 1999a; Nakamura et al., 1997) (Abbildung 1.4). Während sich am C-Terminus des Proteins das aktive Zentrum befindet, sind im Bereich des N-Terminus spezielle Aminosäuresequenzen für die Verankerung der RNA-Matrize und deren Stabilisierung während der kontinuierlich ablaufenden DNA-Synthese zuständig (Bryan et al., 2000). Die RNA-Matrize hat zwei Hauptdomänen: Am 5 -Ende befindet sich die Matrize zum Anheften der Telomer- Wiederholungen. Diese bindet an die katalytische Untereinheit htert (Theimer und Feigon, 2006). Die 3 -Domäne enthält ein H/ACA-Motif, an das z.b. Dyskerin und weitere Proteine binden, was zur Stabilisierung von hter beiträgt (Fu und Collins, 2003). Außerdem konnte gezeigt werden, dass die Sequenz der RNA-Matrize für die Telomerverlängerung von Bedeutung ist. RNA-Mutationen führten zu einer Veränderung der Telomersequenz, was wiederum die Aktivität der Telomerase und die DNA-Synthese beeinflusste (Greider und Blackburn, 1996). Die htert-untereinheit ist für die eigentliche Enzymaktivität verantwortlich und somit für die Verlängerung der Telomere. Der Enzymkomplex assoziiert mit der Einzelstrang-DNA am 3 -Ende der Telomere, die die Sequenz TTAGGG enthalten, wobei die Bindung durch kürzere Telomere signifikant verstärkt wird (Ouellette et al., 2000; Westin et al., 2007). Abbildung 1.4 zeigt schematisch, wie die RNA-Matrize mit dem 3 -Ende der Telomersequenz interagiert und diese durch Anhängen von freien Nukleotidtriphosphaten verlängert. Die Telomerase bewegt sich nach vollzogener Synthese eines Hexamers entlang eines DNA-Primers weiter, wodurch es zu einem kontinuierlichen Verlängern der Telomere 5
13 1 Einleitung kommt (Greider, 1991). Die Besonderheit dieses Enzyms ist, dass die RNA in den Enzymkomplex der Telomerase schon integriert ist. htert hter Dyskerin TTAGGGTTAGGGTTAGGGTTAG AATCCCAA CAAUCCCAAUC 5 Verlängerung Dyskerin TTAGGGTTAGGGTTAGGGTTAG AATCCCAA CAAUCCCAAUC 3 5 Translokation Dyskerin TTAGGGTTAGGGTTAGGGTTAGGGTTAG AATCCCAA CAAUCCCAAUC 3 5 Verlängerung 3 Abb. 1.4 Vereinfachte schematische Darstellung der Telomerase Die Telomerase ist eine reverse Transkriptase, die Telomer-Wiederholungen an die Chromosomenenden heftet. Sie besteht aus einer katalytischen Untereinheit htert, aus einer Telomerase-RNA (hter), die als Matrize für Telomeradditionen dient, aus dem Protein Dyskerin und weiteren Proteinen (nicht dargestellt). Die RNA-Matrize interagiert mit dem 3 -Ende der Telomersequenz und verlängert diese durch Anhängen von freien Desoxynukleotidtriphosphaten. Abbildung verändert nach Telomeraseaktivierung führt zur Immortalisierung von normalen Zellen Während der malignen Transformation stellt die Aktivierung der Telomerase und somit die Immortalisierung von Zellen, einen wichtigen Schritt dar. Diese Erkenntnis machte man sich zu Nutze, um normale Zellen zu immortalisieren: Wird in diese durch Transduktion das htert-gen eingebracht, kann eine Telomeraseaktivität induziert werden, was mit einer Verlängerung der Telomere einhergeht (Bodnar et al., 1998). In fast allen Zellarten führt die Stabilisierung der Telomere durch Überexpression von htert zur Immortalisierung dieser Zellen (Bodnar et al., 1998; Harada et al., 2003; Meyerson, 1998; Vaziri und Benchimol, 1998). Dagegen führte ein Telomerase-knock-out der RNA-Matrize in Mäusen zu einer kontinuierlichen Telomerverkürzung und einem frühzeitigen fast vollständigen Verlust von Telomeren (Blasco et al., 1997). Mäuse, die homozygot für mter-/- waren, entwickelten vor 6
14 1 Einleitung allem in Geweben mit hoher Proliferationsrate Defekte, wie z.b. im hämatopoetischen System, dem Magen-Darm-Trakt und in Hautzellen (Rudolph et al., 1999). Die Telomeraseexpression wird während der Immortalisierung hochreguliert (Counter et al., 1992) und es konnte gezeigt werden, dass 85% der humanen Tumore Telomerase-positiv sind (Shay und Bacchetti, 1997), wobei die restlichen 15% über einen alternativen Telomererhaltungsmechanismus ihre Telomere stabil halten Der alternative Mechanismus zur Verlängerung der Telomere In Hefemutanten, denen das Enzym Telomerase fehlte, konnte beobachtet werden, dass diese trotzdem überleben konnten. Überlebende Hefezellen nutzten dafür offenbar Telomerase-unabhängige Telomererhaltungsmechanismen, die Rad50- und Rad51-abhängig sind (Le et al., 1999; Teng und Zakian, 1999). In Typ I-überlebenden Hefezellen, kam es zu einer Amplifikation von sub-telomerischen DNA-Sequenzen und kurzen Telomeren, die als Y bezeichnet wurden. Typ II-überlebende Hefezellen benötigen Rad50p und zeigten lange und heterogene Telomere (Lundblad und Blackburn, 1993). Auch in manchen humanen Zelllinien konnte beobachtet werden, dass ihre Telomerlänge über viele hundert Passagen auch in Abwesenheit von Telomerase erhalten bleibt (Bryan et al., 1995). Ähnliche Beobachtungen wurden nach Transduktion humaner Fibroblasten mit der Early Region des SV40-Virus gemacht, von denen einige Zellen auch ohne nachweisbare Aktivierung der Telomerase immortal wurden (Bryan und Reddel, 1997). Man geht davon aus, dass diese Telomerasenegativen Tumorzellen über mindestens einen alternativen Telomererhaltungsmechanismus (Alternative Lengthening of Telomeres; ALT) verfügen (Bryan und Reddel, 1997). Die Aktivierung dieser ALT-Mechanismen konnte zunächst in humanen Tumoren, immortalisierten Zelllinien oder in Telomerase-negativen Mauszelllinien (Bryan et al., 1997; Bryan et al., 1995; Niida et al., 2000), also in nicht normalen Zellen gefunden werden. Tumore, die einen ALT- Mechanismus aufweisen, sind hauptsächlich mesenchymalen Ursprungs wie z.b. Weichteilsarkome, Astrozytome und Osteosarkome (Aogi et al., 2000; Bovee et al., 2001). Dass der ALT-Mechanismus in Tumoren mesenchymalen Ursprungs gehäuft auftritt, mag daran liegen, dass mesenchymale Zellen einen langsameren Zellumsatz als epitheliale Zellen haben und so die Telomerase dort stärker unterdrückt wird (Henson et al., 2002) Charakteristika von ALT-positiven Zellen In den meisten Telomerase-positiven Zellen sind die Telomerlängen relativ homogen mit einer Durchschnittslänge von 20 kbp. In den meisten Telomerase-positiven Tumorzellen sind die Telomere ebenso relativ homogen jedoch mit einer kürzeren Durchschnittslänge von nur 10 kbp (Bryan et al., 1995; Park et al., 1998). Dagegen sind die Telomere in ALT-Zellen zum Teil sehr heterogen und haben eine längere Durchschnittslänge (20 kbp). Sie reichen zudem von 7
15 1 Einleitung weniger als 3 kbp bis über 50 kbp (Bryan et al., 1997; Bryan et al., 1995; Murnane et al., 1994; Opitz et al., 2001). Diese Telomerlängen lassen sich mit TRF (Terminal Restriction Fragments) - Analysen und nachfolgender Southern Blot-Analyse messen. Dabei wird jedoch eine gesamte Zellpopulation untersucht. Zum anderen können die Telomerlängen mit Q-FISH-Analysen (quantitative Fluoreszenz-in-situ-Hybridisierung) bestimmt werden (Bryan et al., 1995). Der Vorteil der Q- FISH-Analysen besteht darin, dass die Telomerlängen auf chromosomaler Ebene an einzelnen Metaphasen bestimmt werden können. Mit Hilfe von Q-FISH-Analysen lassen sich somit extrem lange und extrem kurze bzw. Telomer-freie Enden an Einzelzellen unterscheiden, was charakteristisch für ALT ist. Die Fluoreszenzstärke korreliert hierbei mit der Telomerlänge. Ein weiteres Merkmal ALT-positiver Zellen ist das erhöhte Auftreten von extrachromosomaler Telomer-DNA in Form von Doppelstrang-Telomer-circle (t-circles) (Cesare und Griffith, 2004; Wang et al., 2004), Einzelstrang-circles (C- oder G-circles) (Henson et al., 2009; Nabetani und Ishikawa, 2009) und linearer Doppelstrang-DNA (Ogino et al., 1998; Tokutake et al., 1998). Es wird angenommen, dass t-circles durch Auflösen der T-Loop-Struktur durch Rekombinationsenzyme zustande kommen, und so in freie t-circles und verkürzte Telomere resultieren (Wang et al., 2004). Doppelstrang-t-circles scheinen im ALT-Mechanismus und in normaler Telomerbiologie von Bedeutung zu sein (Tomaska et al., 2009). Spezifischer als t-circles scheinen in ALT-Zellen C-circles zu sein. So konnte vor Kurzem gezeigt werden, dass beim Auftreten des ALT-Mechanismus vermehrt C-circles, bestehend aus teilweiser Doppelstrang- DNA, gefunden wurden, die aus einem vollständigen C-reichen Strang und aus einem unvollständigen G-reichen Strang bestanden. In ALT-Zellen kommen diese C-reichen Stränge bis zu 1000 fach erhöht vor (Henson et al., 2009). Diese extrachromosomale DNA, sowie auch chromosomale DNA findet man in so genannten Promyelozytischen Leukämie Kernkörperchen (PML-Bodies; promyelocytic leukemia nuclear bodies), die in vielen Geweben auftreten und denen eine wichtige Rolle z.b. bei der Zellzyklusregulation, Differenzierung, Seneszenz und Apoptose zugesprochen wird (Maul et al., 2000; Ruggero et al., 2000). PML- Bodies sind somit ein weiteres Charakteristikum von ALT-Zellen und werden als ALTassoziierte PML-Bodies (APBs) bezeichnet (Yeager et al., 1999). APBs sind subnukleäre Strukturen, bei denen das PML-Protein mit Telomer-DNA, Telomer-bindenden Proteinen und u.u. mit Proteinen, die an der DNA-Synthese und Rekombination beteiligt sind, kolokalisiert (Yeager et al., 1999). Letztere sind die Proteine Mre11, Rad50 und Nbs1, die den so genannten MRN-Komplex bilden (Wu et al., 2000; Zhu et al., 2000). APBs treten auf, sobald der ALT-Mechanismus aktiv ist und sie verschwinden wieder wenn der Mechanismus in somatischen Zellhybriden unterdrückt wird (Perrem et al., 1999). Eine Funktion, die den APBs zugeschrieben wird, ist, dass sie Proteine, die für den ALT-Mechanismus relevant sind, 8
16 1 Einleitung rekrutieren oder modifizieren (Henson et al., 2002). In den meisten Zellen erscheinen APBs in der späten S/G2/M-Phase des Zellzyklus, da zu dem Zeitpunkt die homologe Rekombination am aktivsten ist (Wu et al., 2000). Jedoch wurden mittlerweile auch ALT-positive Zellen ohne das Auftreten von APBs beschrieben (Cerone et al., 2005; Marciniak et al., 2005). Ein weiteres Charakteristikum von ALT-positiven Tumoren ist, dass sie zum Teil Chromosomen enthalten, die nicht mehr durch eine T-Loop geschützt werden und somit vermehrt eine chromosomale Instabilität aufweisen, was schließlich zu breaking-fusion-bridge-zyklen führen kann (Cheung und Deng, 2008; Scheel et al., 2001) ALT-Mechanismus und Telomerrekombination Es wird angenommen, dass ein Teil der ALT-Telomererhaltungsmechanismen auf homologer Rekombination beruhen. Erste Hinweise darauf, dass Rekombination nicht nur in Telomerasenegativen Hefen, sondern auch in humanen ALT-positiven Tumorzellen eine Rolle spielt, zeigten Untersuchungen an einzelnen Chromosomen in immortalen Telomerase-negativen Zellen. Sowohl eine auffallend schnelle Abnahme, als auch eine schnelle Zunahme an Telomersequenzen an den Chromosomen wurde auf einen Austausch von Telomeren durch Rekombination zurückgeführt (Murnane et al., 1994). Weiter konnte eine Telomerverlängerung über Rekombination an der ALT-positiven Zelllinie GM847 demonstriert werden. Dafür wurde ein Marker in die Telomersequenz integriert, der nachweislich in andere Telomere kopiert wurde. Eine Kopie des Markers konnte jedoch in keiner Telomerasepositiven Zelllinie nachgewiesen werden, auch nicht, wenn dieser lediglich in das Sub- Telomer integriert wurde (Dunham et al., 2000). Des Weiteren konnten quantitative Analysen in ALT-positiven Zellen einen post-replikativen Telomeraustausch zwischen Chromosomen und Schwesterchromatiden mit Hilfe des Telomer-Chromosomen-Orientierungs-FISH (CO- FISH) zeigen (Londono-Vallejo et al., 2004). Auch das Mitwirken von Rad51 in ALT-positiven Hefezellen und das Auftreten von Proteinen in manchen APBs, die an der Rekombination beteiligt sind, weisen darauf hin, dass dieser Mechanismus rekombinationsabhängig sein könnte. Es werden zurzeit verschiedene Arten der Rekombination in ALT-Zellen diskutiert: (1) ein post-replikativer Telomeraustausch zwischen Chromosomen oder Schwesterchromatiden, und (2) das Kopieren von Telomeren über homologe Rekombination (Cesare und Reddel, 2008) Post-replikativer Telomeraustausch zwischen Chromosomen und Schwesterchromatiden Dieses Modell basiert auf Beobachtungen, dass ein Telomeraustausch zwischen Chromosomen und Schwesterchromatiden in einer viel höheren Frequenz in ALT-positiven Zellen auftrat, als in Telomerase-positiven Zellen und normalen Zellen (Bechter et al., 2004; 9
17 1 Einleitung Londono-Vallejo et al., 2004). Dies führte zu der Hypothese, dass ein ungleicher Austausch von Telomeren zwischen Schwesterchromatiden zu einer Tochterzelle führt, die verlängerte Telomere aufweist und zu einer zweiten Tochterzelle mit kürzeren Telomeren (Bailey et al., 2004). Das würde eine unbegrenzte Proliferation der einen Zellpopulation begünstigen, vorausgesetzt alle verlängerten Telomere gelangen in eine Tochterzelle. Die andere Tochterzelle würde über alle verkürzten Telomere verfügen, die eine geringere Proliferationskapazität aufweisen würde (Blagoev und Goodwin, 2008; Muntoni und Reddel, 2005) (Abb. 1.5). Bisher wurde angenommen, dass es bei der Mitose zu einer zufälligen Verteilung der Chromosomen kommt. Die Existenz eines Mechanismus, bei dem die Telomeraufteilung nicht zufällig stattfindet, ist bis jetzt noch nicht etabliert und bleibt somit hypothetisch, obwohl es einen Hinweis für eine signifikante nicht zufällige Schwesterchromatidaufteilung in einigen Mauszellen gibt (Falconer et al.). DNA-Replikation Ungleicher Telomer- Schwesterchromatidaustausch Regulierte Chromatidtrennung Erhöhte Proliferationskapazität durch verlängerte Telomere Geringere Proliferationskapazität durch verkürzte Telomere Abb. 1.5 Ungleicher Telomer-Schwesterchromatidaustausch Auf die semi-konservative DNA-Replikation kann eine homologe Rekombination zwischen zwei Schwesterchromatiden folgen und somit zum post-replikativen Telomeraustausch führen. Dabei könnte eine Tochterzelle mit verlängerten Telomeren entstehen und einer erhöhten Proliferationskapazität und eine zweite Tochterzelle mit kurzen Telomeren, die eine geringere Proliferationskapazität aufweist. Abbildung verändert nach Cesare 2010 (Cesare und Reddel) Kopieren von Telomeren über homologe Rekombination Eine zweite Hypothese ist, dass im ALT-Mechanismus über das Kopieren von Telomer-DNA- Sequenzen, die als Matrize für das neusynthetisierte Telomer dienen, Telomere aufrecht erhalten werden (Dunham et al., 2000; Henson et al., 2002) (Abbildung 1.6). ALT-positive Zellen waren in der Lage einen Marker, der in die Telomersequenz integriert wurde, von einem Chromosom auf ein anderes zu kopieren (Dunham et al., 2000) (Abbildung 1.6). In 10
18 1 Einleitung diesem zurzeit bevorzugten Modell (Cesare und Reddel) stellt die DNA-Matrize nicht notwendigerweise ein anderes Chromosom dar. Als Matrize könnten ebenso, neben einem Schwesterchromatid (Abbildung 1.7 C), die eigene T-Loop-Struktur (Abbildung 1.7 A) sowie lineare extrachromosomale Telomer-DNA (Abbildung 1.7 B) und t-circles als roll and circle - Mechanismus (Abbildung 1.7 D) in Frage kommen (Cesare und Reddel). Intertelomerische Stranginvasion und Telomerverlängerung Synthese des zweiten Strangs und Erhalt von verlängerten Telomeren Abb. 1.6 Schematische Darstellung der homologen rekombinationsabhängigen DNA-Replikation Hierbei invadiert der 3 -Überhang des kurzen Telomerstrangs in ein benachbartes Telomer, gefolgt von der Extension des invadierten Strangs durch die DNA-Polymerase. Der C-reiche Strang wird aufgefüllt und somit das Telomer verlängert. Abbildung nach Cesare, 2010 (Cesare und Reddel). Die Möglichkeit, dass in ALT-Zellen der Mechanismus des roll and circle -Mechanismus zur Telomerverlängerung Verwendung findet, unterstützen die Beobachtungen in Hefen, in denen dieser Prozess mit interchromosomaler Rekombination mit einem roll-and-spread -Mechanismus kombiniert wird (Tomaska et al., 2009). Des Weiteren konnte gezeigt werden, dass C- circles in vitro ein exzellentes Substrat für die Amplifizierung durch das Modell des rolling circle darstellen (Henson et al., 2009) und man spekuliert, dass diese ebenso als Substrat für Telomerverlängerung in ALT-Zellen in vivo dienen. Das Anheften eines G-reichen Überhangs an eine Einzelstrang-Region im C-circle, mit anschließender DNA-Polymerisierung vom Chromosomenende würde eine schnelle Synthese der G-reichen Telomer-DNA begünstigen (Cesare und Reddel). 11
19 1 Einleitung A C B D Abb. 1.7 Verschiedene Darstellungen der homologen rekombinationsabhängigen DNA-Replikation Entweder dient die eigene T-Loop-Struktur als Matrize zur Telomerverlängerung (A), lineare extrachromosomale DNA (B), das Telomer eines Schwesterchromatids (C) oder zirkuläre extrachromosomale DNA (D). Der grüne Pfeil zeigt die mutmaßliche Schnittstelle des C-reichen Strangs. Abbildung nach Cesare, 2010 (Cesare und Reddel) Telomererhaltungsproteine in ALT-Zellen Die Komponenten des MRN-Komplexes Mre11 (meiotic recombination 11), Rad50 und Nbs1 waren die ersten Proteine, die als notwendige Proteine im ALT-Mechanismus beschrieben wurden (Wu et al., 2000). Der MRN-Komplex ist normalerweise während der S- und G2- Phase an den Telomeren lokalisiert, indem er direkt mit TRF2 interagiert und so zur T-Loop- Formation von neu replizierten Telomeren beiträgt (Verdun et al., 2005). Beim ALT- Mechanismus könnte es zum Verlust der komplexen Kontrolle des Telomer-Cappings kommen, was zu einer Telomer-spezifischen Erhöhung der homologen Rekombination führen kann (Cesare und Reddel, 2008). In der G1-Phase des Zellzyklus ist die T-Loop-Struktur ausgebildet, muss jedoch in der S-Phase für die Replikation geöffnet werden. In der G2- Phase wird die T-Loop Struktur wieder hergestellt, wobei die Proteine TRF1, TRF2, Tin2, Rad51 und Rad52 sowie MRN eine wichtige Rolle spielen. Kommt es zur Deregulierung des Telomer-Cappings am eigenen Telomer, kann dies zu Telomerrekombination zwischen verschiedenen Telomeren führen. Außerdem können kritisch kurze Telomere entstehen, die anfällig für Rekombinationen sind oder die ungeschützten Enden können einen Interaktion mit APBs eingehen (Cesare und Reddel, 2008) (Abbildung 1.8). 12
20 1 Einleitung Deregulation in HR-vermitteltem Capping: T-Loop Auflösung Rekombination zw. verschiedenen Telomeren anstatt am eigenen Telomer M ALT-assoziierte Defekte Capping durch homologe Rekombination TRF1, TRF2, Tin2, Rad51, Rad52, MRN G2 G1 S Defekte im Telomer-Capping in ALT-Zellen: Kritisch kurze Telomere, die anfällig für Rekombination sind oder mit APBs interagieren Öffnen der T-Loop- Struktur während der DNA-Replikation Abb. 1.8 Deregulierung der T-Loop-Bildung während des Zellzyklus kann zum Entstehen des ALT-Mechanismus führen. In der G1-Phase sind die Telomere durch die Ausbildung der T-Loop-Struktur vor Doppelstrangbrüchen (DSB) geschützt. In der S-Phase, wenn die DNA repliziert wird, muss die T-Loop-Struktur aufgebrochen werden. Letztendlich wird in der G2-Phase die T-Loop-Struktur wieder hergestellt. Dieser Prozess ist streng reguliert und kann durch Deregulation zum Auftreten von ALT führen. Dies passiert entweder durch eine T-Loop-Auflösung, einer Rekombination zwischen verschiedenen Telomeren anstatt am eigenen Telomer oder durch die Interaktion mit APBs. Abbildung nach Cesare, 2008 (Cesare und Reddel, 2008). All diese Modelle zeigen, dass es sich bei ALT um einen sehr komplexen Mechanismus handelt, der im Detail noch nicht hundertprozentig verstanden wird. Hinter dem ALT- Mechanismus werden sich wahrscheinlich mehrere Mechanismen verbergen (Johnson et al., 2005). Dass unterschiedliche ALT-Mechanismen möglich sind, wurde auch in einer weiteren Studie gezeigt, in der nach spontaner Inaktivierung der Telomerase Fibroblasten weiter proliferieren (Kumakura et al., 2005). Jedoch fehlten diesen Zellen ALT-spezifische Eigenschaften wie heterogene Telomerlängen und APBs. Beim rekombinationsbasierten ALT- Mechanismus werden jedoch in diesen APBs die Rekombinationsproteine rekrutiert. Daher erhält eventuell nur eine Subgruppe der ALT-positiven Zellen ihre Telomere über Rekombination und die restlichen über einen rekombinationsunabhängigen Mechanismus. Es gibt also wahrscheinlich nicht den einen ALT-Mechanismus, sondern verschiedene ALT- Mechanismen. Alle ALT-Zellen weisen jedoch als einzige gemeinsame Eigenschaft den Erhalt von Telomeren, trotz fehlender Telomeraseaktivität, auf. Somit ist der nachgewiesene Erhalt der Telomerlängen mit gleichzeitigem Fehlen der Telomeraseaktivität im TRAP-Assay der bisher einzige valide Nachweis eines ALT-Mechanismus (Henson und Reddel). 1.3 Bedeutung der Telomererhaltung in der Tumorgenese Normale humane Zellen besitzen, bedingt durch den Verlust von Telomersequenzen nach jeder DNA-Replikation, wie beschrieben, eine begrenzte Lebensspanne (Harley et al., 1990). 13
21 1 Einleitung Eine kritisch kurze Telomerlänge veranlasst die Zellen in das Stadium der Seneszenz überzugehen und letztendlich den Weg der Apoptose einzuschlagen (Hahn und Meyerson, 2001). Dieser Mechanismus trägt zum Schutz des Gesamtorganismus bei (Wright und Shay, 2001). Frühe Studien zur htert-expression zeigten, dass die Telomerase in humanem somatischem Gewebe unterdrückt wird, jedoch in Keimzellen und Tumorzellen robust exprimiert wird (Kim et al., 1994). Keimbahnzellen und embryonale Zellen jedoch sind in der Lage ihre Telomerlängen so stabil zu halten und auch nach Klonen adulter Zellkerne deren verkürzte Telomere wieder zu verlängern (Schaetzlein et al., 2004). In normalen Zellen konnte zunächst auch keine Aktivität der Telomerase festgestellt werden (Dahse et al., 1997). Sensitivere Nachweismethoden zeigten aber auch in primären humanen Zellen ein geringes Maß an Telomeraseaktivität, v.a. in stark proliferierenden Geweben mit hohem Erneuerungspotential, wie z.b. Knochenmark, Haut, Gastrointestinaltrakt und aktivierten Lymphozyten (Forsyth et al., 2002; Liu et al., 1999; Yui et al., 1998). Diese Telomeraseaktivität alleine reicht jedoch nicht aus, die Telomerlängen stabil zu halten, da sich die Telomerlängen weiterhin verkürzten (Allsopp et al., 2001; Son et al., 2000). Masutomi et al konnten sogar eine htert- Expression und Telomeraseaktivierung in frühen Passagen humaner Fibroblasten v.a. in der S-Phase nachweisen (Masutomi et al., 2003). Dies zeigte, dass die Regulation von htert und Telomerase in normalen Zellen Zellzyklus-abhängig und wichtig für die Proliferation von normalen humanen Zellen ist (Masutomi et al., 2003). Einer der ersten Hinweise auf eine Rolle der Telomere in der Tumorentstehung wurde an Untersuchungen an primären humanen Fibroblasten erbracht. Diese primären humanen Zellen erreichten ein limitiertes Replikationspotential nach ca Populationsdopplungen bevor sie in das Stadium der Seneszenz übergingen (Hayflick und Moorhead, 1961). Im Gegensatz dazu teilten sich etablierte Tumorzellen unbegrenzt in Kultur. Beobachtungen an primären Zellen und Tumorzellen wiesen darauf hin, dass dieser Unterschied mit der Verkürzung der Telomere einherging. In normalen primären Zellen kam es nach jeder Zellteilung zu einem Verlust von Telomersequenzen, in Tumorzellen hingegen nicht (Harley et al., 1990). Ein Telomer-Verlust nach jeder Zellteilung limitierte die Lebensspanne von Zellen in Kultur (Klapper et al., 2001). Tumorzellen erreichen jedoch über eine Stabilisierung der Telomere durch die Aktivierung von Telomerase oder einem ALT-Mechanismus ein Stadium der Immortalität und überwinden so das Seneszenzstadium. Immortalisierung ist eine wichtige Vorraussetzung für ein ungehindertes Tumorwachstum und somit ein essentieller Schritt in der malignen Transformation von Zellen (Hahn und Weinberg, 2002)(Abbildung 1.3). Die Immortalisierung ermöglicht so eine ungehinderte Proliferation von Tumorzellen und erlaubt es auf dem Weg der malignen Transformation weiter genetische Alterationen zu akkumulieren. 14
22 1 Einleitung Zur Stabilisierung der Telomere in Tumorzellen kommen prinzipiell beide Telomererhaltungsmechanismen in Frage. Ungefähr 85% zeigen eine erhöhte Telomeraseaktivität (Kim et al., 1994) und 15% einen alternativen Telomererhaltungsmechanismus. Dass die Mehrheit der Tumore während der malignen Transformation Telomerase aktivieren, mag daran liegen, dass diese mehrheitlich epithelialen Ursprungs sind, also Karzinome, und das Epithel, aufgrund seiner gering ausgeprägten physiologischen Telomeraseaktivität einer Reaktivierung des Enzyms weniger entgegenzusetzen hat (Neumann und Reddel, 2002). Dagegen aktivieren Tumore mesenchymalen Ursprungs häufig ALT zur Telomererhaltung (Bryan und Reddel, 1997). Möglich wäre auch, dass der ALT-Mechanismus schon während der malignen Transformation eine wichtige Rolle einnimmt. In einem Karzinogenesemodell lassen sich Plattenepithelzellen oral-ösophagealen Ursprungs nach Inaktivierung des Tumorsuppressorgens p53 und der Überexpression des Zellzyklusproteins Cyclin D1 über ALT immortalisieren (Opitz et al., 2001). Eine zusätzliche Überexpression des epidermalen Wachstumsfaktorrezeptors (EGFR) führte zu einer in vitro- Transformation dieser Zellen mit dann einhergehender Aktivierung der Telomerase (Goessel et al., 2005; Heeg et al.). Dies führte zur Arbeitshypothese, dass in der frühen Karzinogenese generell die Rekrutierung des ALT-Mechanismus zur Immortalisierung notwendig ist und erst in der späteren malignen Transformation Telomerase über verschiedene Mechanismen aktiviert wird (Goessel et al., 2005; Heeg et al.). Auch Daten anderer Arbeitsgruppen unterstützen die Hypothese einer frühen Rolle von ALT in der Tumorgenese (Rudolph et al., 1999). Da ca. 85% der immortalen Zellen und Tumorzellen das Enzym Telomerase aktivieren und es somit eine zentrale Bedeutung während der Karzinogenese einnimmt, wird es zu einem attraktiven Ziel bei der Entwicklung von neuen Krebstherapeutika. So führt zum Beispiel die Hemmung von htert durch Überexpression von dominant-negativen htert-mutanten zu Verkürzung der Telomere in Tumorzellen und schließlich sogar zum Zellzyklusarrest oder Apoptose (Hahn et al., 1999; Zhang et al., 1999). Ähnliche Ergebnisse konnten auch nach lentiviraler Überexpression von mutierten RNA-Matrizen und sirna komplementär zur hter- Sequenz gezeigt werden (Li et al., 2004). Diese Experimente zeigen, dass eine Inhibition der Telomerase theoretisch ein funktioneller Angriffspunkt in der Tumortherapie darstellen könnte. Einige Telomeraseinhibitoren sind derzeit schon in verschiedenen klinischen Phasen, wie z.b. Enzyminhibitoren oder verschiedene aktive Telomeraseimmuntherapien (Harley, 2008). Hierbei stellt sich die Frage, ob Telomerase und ALT in ein und demselben Tumor oder sogar in derselben Zelle vorkommen können. Es wäre denkbar, dass durch die Inhibition der Telomerase der Selektionsdruck in Tumorzellen so ansteigt, dass diese einfach auf den alternativen Telomererhaltungsmechanismus wechseln können. Hinweise darauf, dass ALT 15
23 1 Einleitung und Telomerase in derselben Zelle artifiziell funktionieren können, konnte an der ALTpositiven Zelllinie GM847 gezeigt werden. In diesen Zellen konnten die kürzesten Telomere nach exogener Telomeraseexpression verlängert werden und mit Hilfe des TRAP-Assays konnte eine in vitro Telomeraseaktivität nachgewiesen werden. Jedoch behielten diese Zellen weiterhin Merkmale des ALT-Mechanismus wie heterogene Telomerlängen und APBs (Perrem et al., 2001). Des Weiteren wurden Tumore gefunden, die beide Telomererhaltungsmechanismen aufzeigen (Bryan et al., 1997). Jedoch konnte man noch nicht ausschließen, ob es innerhalb eines Tumors verschiedene Subpopulationen gibt, die den einen oder den anderen Telomererhaltungsmechanismus einschlagen. Beobachtungen, dass APBs in der G2- und M-Phase gehäuft auftreten (Grobelny et al., 2000; Wu et al., 2000), jedoch die Telomeraseaktivität in normalen Fibroblasten in der S-Phase auftritt (Masutomi et al., 2003), führten zu der Überlegung, dass die Koexistenz beider Mechanismen Zellzyklus-reguliert sein könnte. Dies konnte an Telomerase-immortalisierten Keratinozyten des Ösophagus bestätigt werden (Von Werder, 2007). Ein hoher Anteil an Zellen befand sich unter exponentiellen Wachstumsbedingungen in der S-Phase; die Telomerlängen waren dabei homogen mit konstanter Telomeraseaktivität. Wurden die Zellen in der G1- bzw. G2-Phase des Zellzyklus durch konfluentes Wachstum oder mit Behandlung von Natrium-Butyrat arretiert, kam es rasch zu heterogenen Telomerlängen und einer deutlich reduzierten Telomeraseaktivität. Darüber hinaus war das Auftreten von APBs in den Zellzyklus-arretierten Zellen deutlich erhöht. Diese Ergebnisse deuten darauf hin, dass beide Telomererhaltungsmechanismen in derselben Zellpopulation existieren, und dass deren Regulation Zellzyklus-abhängig ist (Von Werder, 2007). Es wäre also möglich im Hinblick auf eine Anti-Telomerase-Therapie, dass Telomerasepositive immortale Zellen wie Tumorzellen, die mit Telomeraseinhibitoren behandelt werden, einen alternativen Telomererhaltungsmechanismus aktivieren. 1.4 Das Plattenepithelkarzinom des Ösophagus als Modell zur Untersuchung der Telomerbiologie Die Entwicklung von humanen ösophagealen Plattenepithelkarzinomen ist ein schrittweiser Prozess. Auf eine gesteigerte Proliferation von ösophagealen Plattenepithelzellen (Basalzell- Hyperplasie) folgt eine Dysplasie, ein in situ Karzinom und schließlich ein fortgeschrittenes ösophageales Plattenepithelkarzinom (Lehrbach et al., 2003). Auf diesem Weg kommt es zur Aktivierung von Onkogenen und der Inaktivierung von Tumorsuppressorgenen, die im nachfolgenden näher beschrieben werden: Eine Überexpression des Protoonkogens Cyclin D1, das den Übergang von der G1- in die S-Phase reguliert, ist im ösophagealen Plattenepithelkarzinom die häufigste genetische Alteration (Nakagawa et al., 1995). Neben einer gestörten Regulation des Zellzyklus findet man außerdem eine Überexpression des 16
24 1 Einleitung epidermalen Wachstumsfaktorrezeptors EGFR (Hollstein et al., 1988; Lu et al., 1988; Mandard et al., 2000), der zur Familie der ErbB-Rezeptortyrosinkinasen gehört. Zudem ist das Protoonkogen c-myc in Plattenepithelkarzinomen des Ösophagus häufig amplifiziert (Lu et al., 1988; Mandard et al., 2000) und reguliert unterschiedlichste biologische Funktionen wie Zellproliferation und Differenzierung (Dang et al., 1999). Mechanismen, die zur Inaktivierung von Tumorsuppressorgenen führen, wie z.b. Deletionen von Allelen (LOH) oder Punktmutationen sind ebenso häufige Ereignisse in der Karzinogenese des Plattenepithelkarzinoms des Ösophagus. Dazu zählen vor allem Mutationen im p53 und p16 Tumorsuppressorgen (Gao et al., 1994; Hollstein et al., 1991). In der Arbeitsgruppe wurden verschiedene zelluläre Karzinogenesemodelle etabliert, die in dieser Arbeit verwendet wurden: (1) htert-immortalisierte ösophageale Keratinozyten (EPC-hTERT-Zellen) (Quante et al., 2005), die außer einer Überexpression der Telomerase keine weiteren genetischen Veränderungen aufweisen, und somit eine sehr frühe (prämaligne) Stufe der Plattenepithelkarzinogenese des Ösophagus darstellen sowie (2) generierte Plattenepithelkarzinomzellen, die Cyclin D1, dominant negatives (dn) p53, EGFR und c-myc überexprimieren (Goessel et al., 2005). Diese Zellen sind somit maligne transformiert und stellen eine spätere Stufe der Plattenepithelkarzinogenese des Ösophagus dar. Beide Zelltypen können als genetisch definiert betrachtet werden. 1.5 Das Adenokarzinom des Ösophagus Eine zweite Art des Speiseröhrenkarzinoms ist neben dem Plattenepithelkarzinom das Adenokarzinom. In der Vorstufe, einem Barrett-Ösophagus, wird das Plattenepithel des Ösophagus durch ein metaplastisches Epithelium intestinalen Typs ersetzt. Diese intestinale Metaplasie ist zur Tumorentstehung prädisponiert (Lagergren et al., 1999). Das Adenokarzinom entwickelt sich über diese Barrett-Metaplasie, einer niedriggradigen Dysplasie weiter zu einer hochgradigen Dysplasie und letztendlich zum invasiven Karzinom. Dabei spielen wie im Plattenepithelkarzinom des Ösophagus eine bestimmte Reihenfolge von genetischen Alterationen eine Rolle (Souza et al., 2001). Chromosomale Aberrationen wie Aneuploidie, Verlust der Heterozygotie (LOH), genetische Mutationen und epigenetische Abnormalitäten von Tumorsuppressorgenen sind hier von Bedeutung (Zagorowicz und Jankowski, 2007). Zum Beispiel treten p16-alterationen, wie LOH, Mutationen und Promotormethylierung in 85-90% der Patienten auf (Bian et al., 2002; Eads et al., 2000). Auch die Telomeraseexpression nimmt während der Tumorprogression von einer niedriggradigen zu einer hochgradigen Dysplasie stark zu (Morales et al., 1998). Im Barrett Epithelium wird das Vorhandensein eines Aneuploidiestadiums mit der Progression zur Dysplasie in Zusammenhang gebracht (Reid et al., 2000; Teodori et al., 1998). Somit scheint das 17
25 1 Einleitung Ploidiestadium ein wichtiger Marker für die Progression des Adenokarzinom des Ösophagus zu sein. Des Weiteren findet man im Adenokarzinom zu 85-95% eine p53-alteration (Campomenosi et al., 1996; Muzeau et al., 1996). Im Gegensatz zum Plattenepithelkarzinom des Ösophagus, scheint beim Adenokarzinom das Protoonkogen HER-2/neu anstelle von EGFR eine Rolle zu spielen (Koppert et al., 2005; Rossi et al., 2006). Beide gehören zur Familie der ErbB-Rezeptortyrosinkinasen, jedoch scheint EGFR spezifischer für das Plattenephitelkarzinom und HER-2/neu für das Adenokarzinom des Ösophagus zu sein. Es bestehen zwei Zellmodelle der Barrettkarzinogenese, die in dieser Arbeit Anwendung fanden: (1) CP-A-Zellen, eine Barrettzelllinie, die nur eine p16-deletion besitzt und durch htert immortalisiert wurde (Palanca-Wessels et al., 2003). Diese stellt somit eine frühe (prämaligne) Stufe der Barrett-Adenokarzinogenese dar und kann als genetisch definiert bezeichnet werden. (2) OE19-Adenokarzinomzelllinie (Rockett et al., 1997), eine kommerziell erworbene Zelllinie, die aus einem fortgeschrittenen Adenokarzinom etabliert wurde. Sie ist Telomerase-positiv jedoch sind weitere genetische Alterationen nicht bekannt. Diese Zelllinie stellt somit eine sehr späte Stufe der Barrett-Adenokarzinogenese dar und ist genetisch nicht definiert. 18
26 1 Einleitung 1.6 Zielsetzung Die Immortalisierung von Zellen stellt einen wichtigen Schritt während der malignen Transformation dar. Dieser ist wiederum abhängig vom Erhalt der Telomeren. Zurzeit sind zwei Telomererhaltungsmechanismen bekannt, einerseits das Enzym Telomerase, das in 85% der Tumorzellen aktiv ist, und andererseits ein alternativer Telomererhaltungsmechanismus (ALT), der in ca. 15% der Telomerase-negativen Zellen nachweisbar ist. Bisher ist wenig über die Regulation dieser beiden Mechanismen in einer Zellpopulation und auf Einzelzellbasis bekannt. Es wurde lange davon ausgegangen, dass entweder Telomerase oder ALT in einer Zelle aktiv sein können. Es konnte jedoch in unserer Arbeitsgruppe gezeigt werden, dass Zellen von einem Telomererhaltungsmechanismus auf den anderen wechseln können, und dass dies Zellzyklus-abhängig geschieht (Von Werder, 2007). Da Telomerase in 85% der Tumore aktiv ist, scheint dieses Protein ein guter Angriffspunkt in der Behandlung von Krebspatienten zu sein. Ziel dieser Arbeit war zu untersuchen, ob prämaligne und maligne Stufen der Plattenepithelkarzinogenese des Ösophagus sowie prämaligne und maligne Stufen der Barrett-Adenokarzinogenese von einem Telomererhaltungsmechanismus zum anderen wechseln können, wenn diese einer konsequenten Telomeraseinhibition unterzogen werden. Dies hätte große Auswirkungen auf Telomeraseinhibitoren als Anti- Tumortherapie. Um dies zu untersuchen wurden Telomerase-positive genetisch definierte, immortalisierte, ösophageale Keratinozyten, genetisch definierte, ösophageale Plattenepithelkarzinomzellen, genetisch definierte, immortalisierte Barrettzellen sowie kommerziell erworbene genetisch nicht definierte Adenokarzinomzellen mit verschiedenen genetischen Telomeraseinhibitoren behandelt. Die Inhibitoren enthalten entweder eine mutierte Version der RNA-Matrize oder eine Kombination aus mutierter RNA-Matrize und sirna komplementär zur RNA-Matrize. Diese Konstrukte wurden durch lentivirale Transduktion in die Zellen eingebracht. Es wurde zunächst das Wachstumsverhalten beobachtet und anschließend die Effizienz der Telomeraseinhibition anhand eines TRAP (Telomeric Repeat Amplification Protocol)-Assays auf die Telomeraseaktivität untersucht. Im nächsten Schritt wurden die Telomerlängen an einer Gesamtpopulation mit TRF-Southern Blots und auf Einzelzellbasis mit Q-FISH-Analysen gemessen. Weiterhin wurden eine indirekte Immunfluoreszenz zur Untersuchung von Kolokalisationen von TRF2 und PML-Bodies (APBs) durchgeführt und außerdem wurden die Telomere mit einem Chromosomen-Orientierungs-FISH auf mögliche Schwesterchromatidaustausche untersucht. So sollte aufgezeigt werden, ob verschiedene immortale Zellen sowie invasive Tumorzellen aus dem Ösophagus durch Inhibition der Telomerase über mehr als 50 Passagen auf den ALT-Mechanismus ausweichen können und somit in der Lage sind zu überleben. 19
27 2 Material und Methoden 2 Material und Methoden 2.1 Material Chemikalien Die hier nicht aufgeführten laborüblichen Chemikalien wurden von den Firmen Merck (Darmstadt), Serva (Heidelberg), Roth (Karlsruhe), Sigma-Aldrich (St. Louis, USA), Roche (Mannheim), Invitrogen (Carlsbad, USA), Promega (Madison, USA), AnalaR Normapur (Haasrode, Belgien), Calbiochem (La Jolla, USA) und J.T. Baker (Deventer, Holland) in p.a.- Qualtität bezogen Sonstiges Sonstige Reagenzien 5-Bromo-2 -Deoxyuridine 5-Bromo-2 -Deoxycytidine Anhydrous 6x-Loading-Dye Acrylamid 30%/Bis Lösung Acrylamid 40%/Bis Lösung Agarose Ammoniumpersulfat BioRad Protein Assay Bovines Serum Albumin (BSA) Cold Water Fish Gelatin Dimethylsulfoxid (DMSO) DNA-Sizer III DNA-Leiter 8-48 kb Standard Ethidiumbromid DNA aus Heringssperma Hoechst Hybmix Immersol Karyomax-Colcemid L-Glutamin NEN Blocking Reagenz Nonidet 40 Phosphate buffered saline (PBS) Penicillin/Streptomycin Sigma-Aldrich (St. Louis, USA) MP Biomedicals (Solon, USA) Fermentas (Burlington, Canada) BioRad (Hercules, USA) BioRad (Hercules, USA) Serva (Heidelberg) BioRad (Hercules, USA) BioRad (Hercules, USA) BioLabs (Schwalbach) Sigma-Aldrich (St. Louis, USA) Sigma-Aldrich (St. Louis, USA) PeqLab (Erlangen) BioRad (Hercules, USA) Sigma-Aldrich (St. Louis, USA) Böhringer Mannheim (Mannheim) Invitrogen (Carlsbad, USA) Stratagene (La Jolla, USA) Zeiss (Jena) Invitrogen (Carlsbad, USA) Invitrogen (Carlsbad, USA) DuPont Applichem (Darmstadt) Invitrogen (Carlsbad, USA) Invitrogen (Carlsbad, USA) 20
28 2 Material und Methoden Polybrene (Hexadimethrinbromid) Sigma-Aldrich (St. Louis, USA) SlowFade Antifade Reagenz Invitrogen (Carlsbad, USA) TEMED BioRad (Hercules, USA) T4-Kinase-Puffer Invitrogen (Carlsbad, USA) Trypsin PAA Laboratories GmbH (Pasching, Österreich) Tween 20 BioRad (Hercules, USA) Vectashield + DAPI Vector Laboratories (Burlingame, USA) γ- 32 P-ATP Hartmann Analytic (Braunschweig) Einmalartikel: Plastik und Glaswaren 0,45 µm Spitzenfilter, steril Roth (Karlsruhe) 0,2 ml PCR Gefäß, ultradünn Biozym (Wien, Österreich) 6-Loch Platten Greiner Bio-One (Frickenhausen) 10 cm Zellkulturschale TPP (Trasadingen, Schweiz) 1,5 ml Reaktionsgefäß Eppendorf (Hamburg) 15 ml Reaktionsgefäß Becton Dickinson (Franklin Lakes, USA) 50 ml Reaktionsgefäß Becton Dickinson (Franklin Lakes, USA) Cryo.s TM Einfrierröhrchen Greiner Bio-One (Frickenhausen) Deckglas (24x60mm) Langenbrinck (Emmendingen) Deckglas rund (Ø25 mm) Langenbrinck (Emmendingen) Hybond Nylonmembran GE Healthcare (Giles, UK) Objektträger (76x26 mm) Langenbrinck (Emmendingen) Pasteurpipetten aus Glas Brand (Wertheim) Plastikküvette Starstedt (Nümbrecht) Immuno Blot PVDF-Membran BioRad (Hercules, USA) Röntgenfilm, Kodak X-OMAT Kodak (Rochester, USA) Amersham Hyperfilm GE Healthcare (Giles, UK) Stripetten (5, 10, 25 und 50 ml) Corming Incorporated (Lowell, USA) Whatman Chromatographiepapier Fisher Scientific (Ulm) Fast Read Zählkammer Biosigma (Cona, Italien) Reaktionskits ECL Plus Western Blot Detection Kit Nucleotid Removal Kit EpiTect Bisulfite Kit ProFection Mammalian Transfection System QIAamp DNA-Mini-Kit GE Healthcare (Giles, UK) Qiagen (Hilden) Qiagen (Hilden) Promega (Madison, USA) Qiagen (Hilden) 21
29 2 Material und Methoden TRAPeze Telomerase Detections Kit Millipore (Billerica, USA) Geräte Mikroskope Fluoreszenzmikroskop Axiovert 200 AxioCam MRn Fluoreszenzmikroskop Axioskop 2MOT AxioCam Konfokales Mikroskop Leica DMIRE2 Durchlichtmikroskop Olympus CK30 Zeiss (Jena) Zeiss (Jena) Zeiss (Jena) Zeiss (Jena) Leica Microsystems (Wetzlar) Olympus (Hamburg) Zentrifugen Tischzentrifuge Centrifuge 5415D Kühlzentrifuge Centrifuge 5415R Multifuge 3SR+ Multifuge 3 S-R Eppendorf (Hamburg) Eppendorf (Hamburg) Thermo Fisher Scientific (Waltham, USA) Heraeus (Hanau) Elektrophoresekammern Große DNA-Kammer DNA Sub Cell Mini Trans Blot Cell PerfectBlue Gelsystem Mini M BioRad (Hercules, USA) BioRad (Hercules, USA) PeqLab (Erlangen) Programme LSM 5 Image Browser Zeiss (Jena) Sonstiges FACS Calibur Becton Dickinson (Franklin Lakes, USA) Filmentwicklermaschine Curix 60 AGFA (Köln) Gel-Dokumentationssystem Intas (Göttingen) Geltrockner BioRad (Hercules, USA) Spektrometer Ultraspec 2100 pro Amersham Biosciences (Uppsala, Schweden) Spektrophotometer NanoDrop 1000 Thermo Fisher Scientific (Waltham, USA) Steri-Cycle CO 2 Incubator Thermo Fisher Scientific (Waltham, USA) Sterile Werkbank Clean Air UniTec (Hanau) Wasserbad GLF 1083 Hilab (Karlsruhe) Stratalinker 2400 UV Irradiator Artisan Scientific (Campaign, USA) T1 Thermocycler Biometra (Göttingen) 22
30 2 Material und Methoden Western Blot Kammer Zellsorter MoFlo BioRad (Hercules, USA) Dako (Glostrup, Dänemark) Enzyme Exonuclease III HinfI Pepsin Ribonuclease A RsaI Taq-Polymerase T4-Polynucleotid Kinase Promega (Madison, USA) New England Biolabs (Ipswich, USA) Sigma-Aldrich (St. Louis, USA) Sigma-Aldrich (St. Louis, USA) New England Biolabs (Ipswich, USA) Invitrogen (Carlsbad, USA) Invitrogen (Carlsbad, USA) Zelllinien Zelllinien Eigenschaften Referenz EPC-hTERT Ösophageale Keratinozyten; (Quante et al., 2005) Überexpression von htert OKF6 D1/dnp53/EGFR/myc Orale Keratinozyten; Überexpression (Goessel et al., 2005) von Cyclin D1, dominat-negativem p53, EGFR und c-myc; Telomerase positiv CP-A Barrett-Zellen; Inaktiviertes p16; Überexpression von htert (Palanca-Wessels et al., 1998) CP-B Barrett-Zellen; Inaktiviertes p53; Überexpression von htert (Palanca-Wessels et al., 1998) CP-C Barrett-Zellen; intaktes p16; Überexpression von htert (Palanca-Wessels et al., 1998) OE19 Adenokarzinomzelllinie; Telomerase (Rockett et al., 1997) positiv 293T Mit den Adenovirusgenen E1A und E1B transformierte humane embryonale Nierenfibroblasten (Graham et al., 1977) Plasmide Plasmid Eigenschaften Referenz pcmv R8.91 Expressionsplasmid der HIV-1 (Zufferey et al., 1997) Gag/Pol-Proteinen zur lentiviralen Expression pmd2.g Expressionsplasmid des VSV-G-Env- Proteins für Virusproduktion Didier Trono 23
31 2 Material und Methoden Lentivirale Plasmide: Telomeraseinhibitoren Bezeichnung des Plasmids Name Referenz 1 Backbone: phriu1hter-cmvgfpwsin18; (Li et al., 2004) WT-hTER integriert 2 Backbone: phriu1hter-cmvgfpwsin18 (Li et al., 2004) MT-hTER/AU5 intergiert 3 Backbone: phriu1hter-cmvgfpwsin18 (Li et al., 2004) MT-hTER/47A integriert 6 Backbone: phriu1hter-cmvgfpwsin18 (Li et al., 2004) MT-hTER/AU5+siRNA integriert 7 Backbone: phriu1hter-cmvgfpwsin18 (Li et al., 2004) MT-hTER/47A+siRNA integriert 8 Backbone: phrcmvgfpwsin18 (empty vector) (Li et al., 2004) hter hter hter hter DNA htert A A U C C C T T A G G G DNA htert A U A U A U T A T A T A DNA htert A A A C C C T T T G G G htert A A U C C C U U A G G G sirna WT-hTER MT-hTER/AU5 MT-hTER/47A sirna Abb. 2.1 Schematische Darstellung von WT-hTER, MT-hTER und der zu WT-hTER komplementären sirna. In weiß hervorgehoben die veränderten Nukleotide und die daraus resultierende komplementäre Telomer-DNA in MT-hTER Antikörper Primäre Antikörper Eigenschaften Referenz; Katalognummer Anti-hTERT Monoklonaler Maus IgM gegen die humane Telomerase Reverse Novus Biologicals, Littleton, USA; NB Transkriptase Anti-β-Tubulin Polyklonaler Kaninchen IgG Cell Signaling Technology, Danvers, USA; #2146 Anti-TRF2 Monoklonaler Ziegen IgG Santa Cruz, Santa Cruz, USA; sc9862 Anti-PML Monoklonaler Maus IgG Imgenex, San Diego, USA; IMG124A 24
32 2 Material und Methoden Sekundäre Antikörper Eigenschaften Referenz; Katalognummer ECL-Anti-Kaninchen Meerrettich- Peroxidasekonjugiert Antikörper aus Esel gegen Kaninchen IgG konjugiert mit GE-Healthcare, Little Chalfont, England; NA934V Meerrettich Peroxidase ECL-Anti-Maus Meerrettich- Peroxidasekonjugiert Antikörper aus Esel gegen Maus IgG konjugiert mit Meerrettich- GE-Healthcare, Little Chalfont, England; NA931V Peroxidase Anti-Maus Alexa Fluor Antikörper aus Esel gegen Maus Molecular Probes, Karlsruhe; A IgG konjugiert mit Alexa Fluor 594 Anti-Ziege Cy3 Antikörper aus Esel gegen Ziegen IgG konjugiert mit Cy3 Jackson ImmunoResearch, Suffolk, UK; Telomer-Sonden Sonden Sequenz Referenz TRF-Sonde 5 -CCCTAACCCTAACCCTAA-3 BIG- Biotech, Freiburg Tel-FITC 5 FITC-OO-TTAGGGTTAGGGTTAGGG-3 Eurogentec, Seraing, Belgien Tel-Cy3 5 -Cy3-OO-CCCTAACCCTAACCCTAA-3 PBIO/Biosearch Product, Badford, USA Oligonukleotide Name Sequenz Referenz p16mforward TTA TTA GAG GGT GGG GCG GAT CGC TIB Molbiol, Berlin p16mreverse GAC CCC GAA CCG CGA CCG TTA TIB Molbiol, Berlin p16forward TTA TTA GAG GGT GGG GTG GAT TGT TIB Molbiol, Berlin p16reverse CAA CCC CAA ACC ACA ACC ATA A TIB Molbiol, Berlin 25
33 2 Material und Methoden 2.2 Puffer und Lösungen Zellkultur Zellkulturmedien Medium zur Kultivierung von EPC-hTERT-Zellen: Keratinozytenmedium (Serumfrei, Invitrogen) 37,5 µg/ml Bovine Pituitary Extract 1,26 x 10-3 µg/ml EGF 1 x L-Glutamin 1 x Penicillin/Streptomycin Medium zur Kultivierung von OKF6 D1/p53/EGFR/myc-Zellen: Definiertes Keratinozytenmedium (Invitrogen) 1 ml Supplement 300 µm CaCl 2 1 x L-Glutamin 1 x Penicillin/Streptomycin Medium zur Kultivierung von CP-A-Zellen: Keratinozytenmedium (Serumfrei, Invitrogen) 37,5 µg/ml Bovine Pituitary Extract 1,26 x 10-3 µg/ml EGF 5% (v/v) Fetal Bovine Serum 1 x L-Glutamin 1 x Penicillin/Streptomycin Medium zur Kultivierung von OE19-Zellen: RPMI 1640 (Invitrogen) 10% (v/v) Fetal Bovine Serum 1 x L-Glutamin 1 x Penicillin/Streptomycin 26
34 2 Material und Methoden Medium zur Kultivierung von 293T-Zellen DMEM (Invitrogen) 10% (v/v) Fetal Bovine Serum 1 x L-Glutamin 1 x Penicillin/Streptomycin Einfriermedium Je nach Zelltyp 60 % (v/v) optimales Medium 20 % (v/v) Fetal Bovine Serum 20 % (v/v) Dimethylsulfoxid Molekularbiologie 10% Polyacrylamidgel 25 % Acrylamid 40%/Bis Solution 0,5 x TBE TRAP-Ladepuffer 86 % Formamid 0,025 % (w/v) Bromphenolblau 0,025 % (w/v) Xylencyanol 50 mm EDTA (ph 8,0) 5 x TBE 446,8 mm Trizma Base 442,6 mm Borsäure 10 mm EDTA 50 x TAE-Puffer 2 M Trizma Base 1 M Eisessig 50 mm EDTA ph 8,0 Denaturierungspuffer 1,5 M NaCl 0,5 M NaOH Neutralisierungspuffer 1,5 M NaCl 0,5 M Trizma Base (ph 7,0) 1 mm EDTA (ph 8,0) 27
35 2 Material und Methoden 20 x SSC 3 M NaCl 299 mm Natriumcitrat ph 7,0 2 x SSC 0,3 M NaCl 29,9 mm Natriumcitrat 1 % SDS 0,2 x SSC 30 mm NaCl 2,9 mm Natriumcitrat 1% SDS Biochemie Tabelle 2.1: Pipettierschema der unterschiedlich konzentrierten Polyacrylamidgele Reagenzien 5% Trenngel 8% Trenngel 10% Trenngel 15% Trenngel 5,6% Sammelgel Aqua bidest 4,4 ml 3,5 ml 2,7 ml 1,2 ml 6,6 ml Acylamid 30% 1,6 ml 2,5 ml 3,3 ml 4,8 ml 1,9 ml 1 M Tris-HCl ph 8,7 3,8 ml 3,8 ml 3,8 ml 3,8 ml 1 M Tris-HCl ph 6,8 1,25 ml 10% SDS 100 µl 100 µl 100 µl 100 µl 100 µl 10% APS 100 µl 100 µl 100 µl 100 µl 100 µl TEMED 10 µl 10 µl 10 µl 10 µl 10 µl Sammelgelpuffer 0,5 M Tris-HCl 0,4 % (w/v) SDS 5 x Western Blot Ladepuffer 20% Sammelgelpuffer 3,2%(w/v) SDS 40% Glycerin 0,04% Bromphenolblau 10% SDS 86,8 mm Elektrophoresegrade-SDS (sehr rein) ph 7,2 28
36 2 Material und Methoden 10 x Laufpuffer 247,9 mm Trizma Base 1,92 M Glycin 1% (w/v) SDS 10x Transferpuffer 198,3 mm Trizma Base 1,44 M Glycin 10x TBST 137,9 mm NaCl 2,7 mm KCl 24,79 mm Trizma Base 0,2% Tween-20 ph 8, Indirekte Immunfluoreszenz PBG 0,5% BSA 0,1% Cold Water Fish Gelatin in PBS gelöst Fixierungslösung 3,7% Formaldehyd in PBS verdünnt Permeabilisierungslösung 20% NP40 in PBS verdünnt Fluoreszenz in situ Hybridisierung (FISH) Pepsinlösung 550U/ml verdünnt in PBS Hybridisierungsmix 70% Formamid ultra pure 0,25% NEN Blockier Reagenz 0,5 µg/ml PNA-Sonde (FITC- oder Cy3-markiert) Waschlösung 1 70% Formamid 0,1% BSA 10 mm Tris-HCl ph 7-7,5 29
37 2 Material und Methoden 10x TBS 247,9 mm Trizma Base 362 mm NaCl ph 7,0 Waschlösung 2 1x TBS 0,08% Tween Methoden Zellbiologische Methoden Kultivieren von Zellen Alle Arbeiten mit den verschiedenen Zelltypen wurden unter sterilen Bedingungen an einer Werkbank mit vertikalem laminarem Luftstrom durchgeführt. Die verschiedenen Zellen wurden in dem für sie jeweiligen optimalen Medium unter Punkt kultiviert und beim Erreichen einer Konfluenz von 30-40% passagiert. Dafür wurde das Medium abgenommen, die Zellen mit 5 ml PBS gewaschen und je nach Zelltyp entweder mit einem Trypsin/PBS-Gemisch (1:1) oder mit purem Trypsin von der Platte gelöst. Nach min Inkubation im Brutschrank bei 37 C und 5% CO 2 -Gehalt wurden die gelösten Zellen in 6 ml DMEM aufgenommen und 5 min bei 138 g abzentrifugiert. Anschließend wurde der Überstand verworfen und die Zellen je nach dicke des Zellpellets 1:10 1:80 verdünnt, in frischem Medium aufgenommen und in neue 10 cm Kulturschalen überführt. Die Zellen wurden im Brutschrank bei 37 C und 5% CO 2 in feuchtigkeitsgesättigter Atmosphäre gehalten. Zum Waschen der Zellen wurde lediglich das Medium abgenommen, die Zellen mit 5 ml PBS gewaschen und 8-10 ml frisches für den jeweiligen Zelltypen geeignetes Medium dazugegeben Zellen einfrieren Alle 5-10 Passagen wurde ein Teil der Zellen kryokonserviert. Dazu wurden die Zellen nach Trypsinierung und Abzentrifugation in DMEM in 1 ml optimalem Medium resuspendiert und nach Zugabe von 1 ml Einfriermedium in ein Kryoröhrchen überführt und schließlich durch Einschlagen in ein Papiertuch langsam auf -80 C abgekühlt. Zur längeren Aufbewahrung wurden die Zellen in Flüssigstickstoff gelagert Zellen auftauen Zum Auftauen wurden die Zellen aus dem -80 C Schrank oder Flüssigstickstoff genommen und im 37 C Wasserbad angetaut, bis nur noch ein kleiner Eisklumpen vorhanden war. Die Zellsuspension wurde schnell in ein 15 ml Reaktionsgefäß überführt. Dann wurden vorsichtig 30
38 2 Material und Methoden erst tropfenweise 4 ml DMEM hinzugefügt und dann letztendlich weitere 4 ml DMEM. Anschließend wurden die Zellen 5 min bei 4 C und 138 g abzentrifugiert, der Überstand verworfen und das Pellet in 10 ml frischem Medium resuspendiert und auf eine 10 cm Zellkulturschale überführt und im Brutschrank inkubiert Wachstumskurven erstellen Zunächst wurden konfluente Zellen trypsiniert, anschließend abzentrifugiert und mit einer Zählkammer die Zellzahl bestimmt. Die Zellkonzentration wurde mittels folgender Formel ermittelt: Zellkonzentration (Zellzahl/ml) = mittlere Zahl aus vier ausgezählten Quadraten x 10 4 Nach Bestimmung der Zellzahl wurden 1x 10 5 Zellen pro Zelltyp in einer 10 cm Platte ausgesät und diese bis zu einer Konfluenz von 30% wachsen gelassen. Dann wurden die Zellen erneut abtrypsiniert, abzentrifugiert und gezählt. Die Populationsdopplungen wurden anhand folgender Formel bestimmt: PD= (log(n/n0) : log2) : Anzahl der Tage Wobei N für die neu bestimmte Zellzahl und N0 für die eingesetzte Zellzahl steht. Diese Prozedur wurde alle paar Tage über 3-4 Wochen wiederholt Herstellung von Viruspartikeln Um Plasmid-tragende Lentiviren zu generieren, wurde zunächst die so genannte Packaging -Zelllinie 293T auf einer 10 cm Zellkulturplatte ausplattiert und bis zu einer Konfluenz von 20-30% wachsen gelassen. Dann wurden die Zellen 2x mit 5 ml PBS gewaschen und anschließend 4 h lang mit 5 ml DMEM im Brutschrank inkubiert. Jeweils einer der sechs Lentiviren aus Kapitel wurde zusammen mit dem pmd2.g- und pcmv R8.91-Vektor per Kalziumphosphat Transfektion mit Hilfe des ProFection Mammalian Transfection System (Promega) in die 293T-Zellen eingebracht. Ein Transfektionsansatz setzte sich wie folgt zusammen: 20 µg Lentivektor, 15 µg pcmv R8.91 und 6 µg pmd2.g wurden mit 500 µl 2x HBS und 122 mm CaCl 2 gemischt und mit H 2 O auf 1 ml aufgefüllt und anschließend 10 min bei RT inkubiert. Danach wurden die Röhrchen vorsichtig angeschnippt und der Transfektionsansatz tropfenweise, spiralförmig in die Zellkulturschale gegeben. Nach 12 h Inkubation bei 37 C und 5% CO 2 im Brutschrank wurden die Zellen 2x mit PBS gewaschen. Anschließend wurden 5 ml DMEM auf die Zellen gegeben. Die Virusernten fanden 24, 48 und 72 h nach dem Waschschritt statt. Dafür wurde das DMEM, das die Viruspartikel enthielt, mit einer 10 ml Stripette abgenommen und durch einen 0,45 µm-filter 31
39 2 Material und Methoden in ein 15 ml Reaktionsgefäß filtriert und die Platten mit 5 ml frischem DMEM befüllt. Der Überstand wurde entweder direkt weiterverwendet oder bei -80 C gelagert Lentivirale Transduktion (Spin Transfection) Um eine möglichst hohe Infektionsrate zu erhalten, wurden alle Zellen mittels Infektion durch Zentrifugation (Spin Transfection) mit den Virusüberständen stabil transduziert. Dazu wurden pro 6-Loch Platte 2,5 x 10 5 Zellen in 12 ml Medium ausgesät und über Nacht im Brutschrank bei 37 C und 5% CO 2 inkubiert. Zwei Tage später wurden die Zellen gewaschen und 4 h nach diesem Waschschritt wurde das Infektionsmedium, das sich aus 4 ml Virusüberstand, 2 ml optimalen Medium und 8 µg/ml Polybrene zusammensetzt, auf eine 6-Loch Platte verteilt. Anschließend wurden die Platten 2 h lang bei 800 g und 20 C zentrifugiert und dann über Nacht im Brutschrank bei 37 C und 5% CO 2 inkubiert. Am Folgetag wurde das Infektionsmedium abgenommen und durch 2 ml frisches Medium ersetzt. Einige Tage nach Transduktion, wenn die Zellen dicht genug gewachsen waren, wurden die Zellen an einem Zellsorter MoFlo der Firma Dako (Glostrup, Dänemark) nach GFP sortiert. Dafür wurden sie trypsiniert, in DMEM abzentrifugiert und in 1ml PBS resuspendiert und während dem Sortiervorgang auf Eis gehalten. Nach dem Sortieren wurden die Zellen auf eine frische 10 cm Platte gegeben, welche mit frischem Medium befüllt wurde. Dieser Schritt wurde alle 6-8 Wochen wiederholt Molekularbiologische Methoden Telomere Restriction Amplification Protocol (TRAP) Der TRAP-Assay ist eine sensitive Methode zum Nachweis von Telomeraseaktivität in einem Zellextrakt. Er besteht aus zwei Schritten: im ersten Schritt kommt es, wenn die Telomerase in Zellen aktiv ist, zu einem Anhängen von (TTAGGG)n Sequenzen an ein radioaktivmarkiertes Substrat. Im zweiten Schritt werden die Gesamtkonstrukte mit Hilfe einer PCR und spezifischen Primern amplifiziert (Abbildung 2.2) und auf einem Polyacrylamidgel aufgetrennt, wobei das typische Leitermuster entsteht. Die Intensität und Höhe der detektierten Bandenleiter entspricht der Telomeraseaktivität. Der gesamte experimentelle Aufbau erfolgte nach dem TRAPeze Protokoll der Firma Chemicon. Zur Proteinaufreinigung wurde nach dem Trypsinieren gewonnenes Zellmaterial in 1 ml PBS aufgenommen und bei 5,9 g eine Minute zentrifugiert. Der Überstand wurde verworfen und das Pellet in µl CHAPS-Puffer resuspendiert und 20 min auf Eis inkubiert. Anschließend folgte ein Zentrifugationsschritt für 20 min bei 15,7 g und 4 C. Die Proteinkonzentration wurde nach Bradford, wie unter Punkt beschrieben bestimmt und auf eine Konzentration von 10 ng/µl eingestellt. Der Ansatz wurde gleich weiter- 32
40 2 Material und Methoden verarbeitet oder bei -80 C eingefroren. Aus jedem der eingestellten Proteinextrakten wurden 5 µl entnommen und bei 80 C für 10 min hitzeinaktiviert. Radioaktiv-markiertes Substrat Oligonukleotid 5 γ-32p-aatccgtcgagcagagtt 3 + Telomerase aus Proteinextrakt + dntps + Taq-Polymerase 5 γ-32p-aatccgtcgagcagagtt AGGGTT AGGGTT 3 PCR-Amplifikation AGGGTT AGGGTT AGGGTT 3 AGGGTT AGGGTT AGGGTT AGGGTT 3 Abb. 2.2: Schematische Darstellung des TRAP-Assays Ist Telomerase aktiv, wird im ersten Schritt ein radioaktiv-markiertes Substrat verlängert und im zweiten Schritt durch eine PCR mit spezifischen Oligonukleotiden amplifiziert. Zunächst erfolgte die radioaktive Markierung des Oligonukleotids (TS-Primer) mit einer Sequenz von 5`-AATCCGTCGAGCAGAGTT-3, das als Telomer erkannt wird. Dafür wurden für einen Ansatz von 20 Proben 20 µl des TS-Primers, 10 µl PCR Grade Water, 10 U T4 Polynukleotid Kinase (New England Biolabs) und 1,85 MBq γ- 32 P-ATP gemischt und für 20 min bei 37 C inkubiert. Während diesem Schritt wurde der Austausch der Phosphatgruppe am 5 -Ende des TS-Primers gegen das 32 P aus der γ-position des ATP katalysiert. Anschließend erfolgte eine Hitzeinaktivierung des Enzyms bei 85 C für 5 min. Für den PCR Master Mix wurden 100 µl TRAP-Puffer, 20 µl 50x dntp-mix, 20 µl Primer Mix und 772 µl PCR Grade Water mit den 40 µl radioaktiv-markierten TS-Primer und 40 U Taq-DNA Polymerase (Invitrogen) gemischt. Pro PCR-Ansatz wurden 48 µl Master Mix vorgelegt und jeweils 2 µl Protein (entspricht 20 ng) bzw. hitzeinaktiviertes Protein dazugegeben. Als Positivkontrolle wurde ein Proteinextrakt aus einem Telomerase-positiven Kontrollpellet aus dem Kit eingesetzt und als Negativkontrolle wurden 2 µl CHAPS-Puffer verwendet. Die Verlängerungsreaktion des radioaktiv-markierten TS-Oligonukleotid durch die Telomerase wurde für 30 min bei 30 C in einem T1 Thermocycler (Biometra) durchgeführt. Das PCR- Profil wurde wie folgt verwendet: 33
41 2 Material und Methoden Verlängerungsreaktion 30 min 30 C Denaturierung 5 min 95 C Denaturierung 30 sec 95 C Hybridisierung 30 sec 59 C Elongation 10 min 72 C 27 Zyklen Die PCR-Produkte wurden mit 4 µl TRAP-Ladepuffer versehen und auf einem 10%igen Polyacrylamidgel (12,5 ml 40% Polyacrylamid (19:1), 37,5 ml 0,5x TBE, 500 µl APS, 50 µl TEMED) für 2,5-3 h bei 300 V aufgetrennt. Anschließend wurde das Gel 1-2 h bei 80 C unter Vakuum in einem Geltrockner (BioRad) auf Whatmanpapier getrocknet. Die radioaktiven Banden wurden bei -20 C über Nacht auf einem Röntgenfilm detektiert DNA Extraktion Die DNA-Gewinnung erfolgte mit dem QIAamp DNA Mini Kit (Qiagen) nach Protokoll. Nach Trypsinierung und anschließender Abzentrifugation der Zellen wurden diese in 1 ml PBS resuspendiert und 1 min bei 5,9 g abzentrifugiert. Der komplette Überstand wurde vorsichtig abgenommen und das Pellet in 200 µl PBS resuspendiert. Es folgte eine Zugabe von 20 µl Proteinase K (Qiagen) und dem Puffer AL. Das Gemisch wurde 15 sec gut gemischt und dann bei 56 C 10 min inkubiert. Nach der Inkubation wurden die Reaktionsgefäße kurz abzentrifugiert und jeweils 200 µl 100% Ethanol dazugegeben. Die Ansätze wurden wieder 15 sec gut gemischt und schließlich auf eine QIAamp Mini spin Säule aufgetragen und bei 5,8 g 1 min zentrifugiert. Die Säule wurde in ein neues Auffanggefäß gesetzt, 500 µl AW1-Puffer wurden auf die Säule gegeben und 1 min bei 5,9 g zentrifugiert. Dann wurden 500 µl AW2-Puffer auf die Säule gegeben und 3 min bei 15,7 g zentrifugiert. Zu Elution der DNA wurde die Säule in ein 1,5 ml Reaktionsgefäß gesetzt, µl AE-Puffer dazupipettiert und nach 1 min Inkubation bei RT ein letztes Mal bei 5,8 g zentrifugiert Messung von DNA Die Purin- und Pyrimidinbasen der DNA absorbieren UV-Licht bei 260 nm. Verunreinigungen durch Proteine führen zur Absorption bei 280 nm. Mit folgender Formel wurde die Konzentration berechnet: µg=(a260) x (Verdünnungsfaktor) x (50µg/ml) Verdünnungsfaktor: 200 Multiplikationsfaktor für DNA: 50 34
42 2 Material und Methoden Die Reinheit der DNA-Präparation konnte durch das Verhältnis von A260/A280 bewertet werden. Quotienten von 1,7 2,0 wurden als sauber betrachtet. Die DNA-Messung erfolgte in Quarzküvetten an einem Spektrometer oder an einem Nanodrop Telomere Restriction Fragments (TRF) Der TRF ist eine Methode, die auf einem Southern Blot basiert, und an hand derer die durchschnittliche Telomerlänge eines Zellextrakts bestimmt werden kann. Die dafür erforderliche DNA-Gewinnung wurde wie in Punkt beschrieben durchgeführt. Im nächsten Schritt wurde ein unspezifischer DNA-Verdau mit den Enzymen RsaI und HinfI (New England Biolabs) durchgeführt. Diese beiden Enzyme besitzen Schnittstellen im gesamten Genom, jedoch nicht an den Telomeren, die somit erhalten bleiben. Ein Restriktionsansatz (50 µl) setzte sich wie folgt zusammen: 2 5 µg DNA 1x NEB-Puffer 2 oder 4 10 U RsaI 10 U HinfI Dieser Ansatz wurde über Nacht bei 37 C verdaut. Am Folgetag wurde der Verdau mit Hilfe eines Probegels überprüft. Dafür wurden je 4 µl von verdauter bzw. unverdauter DNA mit 16 µl TE-Puffer (Qiagen) und 4 µl 6x Ladepuffer (Fermentas) versetzt und abwechselnd auf ein 1%iges Agarosegel geladen. Dabei wurde die Agarose im Verhältnis Gewicht zu Volumen (TAE) eingewogen, in der Mikrowelle aufgekocht und mit 0,1 µg/ml Ethidiumbromid versetzt. Dieses Gemisch wurde in horizontalen Gelkammern gegossen und kühlte dort innerhalb einer Stunde ab. Als Laufpuffer diente 1x TAE. Die Auftrennungszeit betrug eine Stunde bei 120 Volt. Bei erfolgreichem Verdau kam es unter UV-Licht zu einem breiten Schmier, wohingegen die unverdauten Proben eine scharf abgetrennte Bande zeigten. Nach diesem Probegel erfolgte die für die Telomerlängenmessung entscheidende Gelauftrennung. Hierfür wurde ein 0,5%iges Agarosegel in einer horizontalen Protean XL- Kammer (BioRad) gegossen. 20 µl der aufzutragenden Proben wurden mit je 4 µl 6x Ladepuffer versetzt. Als Marker wurde der Sizer III (PeqLab) verwendet. Die Laufzeit betrug 14 h bei 40 Volt. Das Gel wurde anschließend unter dem UV-Licht mit angelegtem Lineal als Maßstab betrachtet und fotografiert. Zur Vorbereitung auf den Southern Blot wurde das Gel 15 min in 0,25 M Salzsäure depurinisiert, 40 min in Denaturierungspuffer denaturiert, danach kurz mit A. bidest gespült und abschließend 2x 15 min mit Neutralisierungspuffer gewaschen. Dann wurde ein wannenförmiges Gefäß mit 20x SSC-Puffer befüllt und in dieses ein Sockel gestellt, der aus dem Flüssigkeitsspiegel herausragte. Auf den Sockel wurden je eine Lage längs und quer in Puffer überhängendes und ein auf Gelgröße 35
 und 3 weiter in 20x SSC-Puffer getränkte Whatmanpapiere.")
43 2 Material und Methoden zugeschnittenes Whatmanpapier gelegt und mit 20x SSC-Puffer benetzt. Die nächsten Schichten bildeten das Gel, die auf Gelgröße zugeschnittene Nylonmembran (Hybond, GE Healthcare) und 3 weiter in 20x SSC-Puffer getränkte Whatmanpapiere. Zum Anlegen eines Soges in vertikaler Richtung folgten mehrere Lagen saugfähige, auf Gelgröße angepasste Papiertücher. Die gesamte Konstruktion wurde mit einem Gewicht beschwert und mit Klebeband fixiert. Der Transfer erfolgte für 6-12 h. Gewicht Papiertücher 3 Lagen Whatmanpapier Nylonmembran Agarosegel Längs und quer überhängendes Whatmanpapier Basisblock 20x SSC-Puffer Abb. 2.2 Schematische Darstellung des Southern Blot-Aufbaus In eine mit 20x SSC-Puffer befüllte Wanne wurde ein Sockel gestellt. Der Blot-Aufbau sah wie folgt aus: auf ein längs- und quer hängendes Whatmanpapier gefolgt von dem Agarosegel, folgte eine Nylonmembran und 3 weitere Whatmanpapiere. Ein Stapel Papiertüchern beschwert mit einem Gewicht bildeten den Abschluss des Stapels. Zur Fixierung der DNA auf der Membran wurde diese entweder 1 h bei 80 C im Backofen gebacken, oder in einem UV Stratalinker auf der Membran fixiert. Bevor der eigentliche Hybridisierungsmix auf die Membran gegeben wurde, wurde diese zunächst 30 min bei 37 C im Rollofen mit Hybmix (Stratagene) prä-hybridisiert. Der Hybridisierungsmix setzte sich wie folgt zusammen: 50 pmol DNA- Sonde (TTAGGG) 3 7 µl H 2 O 1x T4-Kinase Puffer 10 U T4 Kinase 1,85 MBq γ- 32 P-ATP Dieser Ansatz wurde 15 min bei 37 C inkubiert und anschließend mit dem Nucleotid Removal Kit (Qiagen) nach Protokoll aufgereinigt: Die radioaktiv-markierte Sonde wurde mit 200 µl PN-Puffer gemischt und auf eine Säule gegeben. Es folgte ein Zentrifugationsschritt für 1 min bei 3,3 g. Zum Waschen der Sonde wurden 500 µl PE-Puffer auf die Säule 36
44 2 Material und Methoden gegeben und ebenfalls 1 min bei 3,3 g zentrifugiert. Dieser Schritt wurde noch mal wiederholt und anschließend wurde die Säule in ein frisches Auffanggefäß gesetzt und bei 15,7 g zentrifugiert. Zum Eluieren wurden 200 µl EB-Puffer zugegeben und für eine Minute bei 15,7 g zentrifugiert. Als nächstes erfolgte eine Zugabe von 300 µl 10 mg/ml Heringssperma und eine Hitzeinaktivierung für 10 min bei 95 C. Nach dem Aufkochen wurde die radioaktivmarkierte Sonde zu dem Hybmix dazugegeben. Die Hybridisierung erfolgte bei 37 C im Rollofen für 4-12 h. Danach wurde die Membran zunächst 30 min mit 2x SSC/0,1% SDS bei 32 C gewaschen und abschließend noch mal 2x 15 min mit 0,2x SSC/0,1% SDS bei 37 C. Die Exposition erfolgte mittels Röntgenfilm (Kodak X-OMAT) über Nacht bei -80 C Methylierungsspezifische PCR (MSP) Die Methylierung der DNA eines Promotorabschnitts führt zur Stilllegung von Genen, die z.b. während der Tumorentwicklung eine wichtige Rolle spielen. Die Methylierung der DNA tritt an Cytosinresten in so genannten CpG-Inseln auf. Der Methylierungsstatus der DNA Sequenz kann durch Bisulfitbehandlung bestimmt werden. Die Inkubation von Ziel-DNA mit Natrium Bisulfit führt zum Austausch von unmethylierten Cytosinresten zu Uracil, wobei die methylierten Cytosine erhalten bleiben. Somit führt die Bisulfitbehandlung zu zwei verschiedenen DNA-Sequenzen von methylierter und unmethylierter DNA: Original Sequenz Nach Bisulfitbehandlung Unmethylierte DNA N-C-G-N-C-G-N-C-G-N N-U-G-N-U-G-N-U-G-N Methylierte DNA N-C-G-N-C-G-N-C-G-N N-C-G-N-C-G-N-C-G-N Im zweiten Schritt wird eine PCR durchgeführt, mit jeweils spezifischen Primern, bei der entweder die methylierten bzw. unmethylierten Produkte amplifiziert werden. Die Bisulfitbehandlung wurde nach Angaben den Herstellers (Qiagen) wie folgt durchgeführt: Es wurden 2 µg DNA mit 85 µl Bisulfitmix und 35 µl DNA Protect Puffer vermischt und der Ansatz mit RNase-freiem Wasser auf 140 µl aufgefüllt. Die Bisulfitumwandlung erfolgte in einer PCR Maschine der Firma Biometra: Denaturierung 5 min 99 C Inkubation 25 min 60 C Denaturierung 5 min 99 C Inkubation 85 min 60 C Denaturierung 5 min 99 C Inkubation 175 min 60 C 37
45 2 Material und Methoden Anschließend wurde die konvertierte DNA mit Hilfe des Epi Tect Bisulfit Kits (Qiagen) aufgereinigt. Dafür wurde die DNA in ein 1,5 ml Reaktionsgefäß überführt und mit 560 µl BL Puffer versetzt. Nach Mischen des Ansatzes wurde die Mixtur auf eine Säule überführt und für eine Minute bei maximaler Geschwindigkeit zentrifugiert. Im Anschluss wurden 500 µl BW-Waschpuffer auf die Säule gegeben und nochmals bei maximaler Geschwindigkeit zentrifugiert. Dann wurden 500 µl BD-Puffer dazugegeben und 15 min bei RT inkubiert. Es wurde nochmals eine Minute bei voller Geschwindigkeit zentrifugiert und die DNA zweimal erneut mit Puffer BW gewaschen. Die DNA wurde mit 20 µl EB-Puffer eluiert, indem sie nochmals bei 13,4 g zentrifugiert wurde. Die Bisulfit-behandelte DNA wurde im nächsten Schritt für die methylierungsspezifische PCR verwendet. Hierfür wurden 100 ng DNA mit 1x Taq-Pol PCR-Puffer, 10 mm dntp Mix und je 300 ng der methylierungsspezifischen- bzw. unmethylierungsspezifischen Primern und mit 1 U Taq Polymerase versetzt. Die PCR wurden wie folgt durchgeführt: Denaturierung 3 min 94 C Denaturierung 30 sec 94 C Hybridisierung 40 sec 65 C Elongation 40 sec 70 C Elongation 10 min 70 C 30 Zyklen Die PCR-Produkte (10 µl PCR-Produkt, 10 µl H2O, 4 µl 6x Loading DYE) wurden anschließend auf einem 6%igen Acrylamidgel aufgetrennt und unter einem UV-Licht betrachtet und dokumentiert Biochemische Methoden Proteinextraktion mit CHAPS-Puffer Um Proteine aus dem Zytoplasma und Zellkern zu erhalten wurden sie Zellen mit CHAPS- Puffer lysiert. Dafür wurden zunächst pro Zelltyp cm Zellkulturschalen, die eine Zelldichte zwischen 30-60% enthielten, abtrypsiniert und mit 6 ml DMEM 5 min bei 5,9 g abzentrifugiert. Der Überstand wurde verworfen und das Pellet in 1 ml PBS resuspendiert und in ein 1,5 ml Reaktionsgefäß überführt und die Zellen bei 5,9 g 1 min abzentrifugiert. Das Pellet wurde entweder bei -20 C eingefroren oder direkt weiterverarbeiten. Bei der Weiterverarbeitung wurde das komplette PBS entfernt und das Pellet je nach Dicke in µl CHAPS-Puffer resuspendiert und 20 min auf Eis inkubiert. Anschließend folgte ein Zentrifugationsschritt 25 min bei 4 C und 15,7 g. Der Überstand, der die Proteine enthielt, wurde in ein neues 1,5 ml Reaktionsgefäß überführt und bei -80 C gelagert. 38
46 2 Material und Methoden Proteinbestimmung nach Bradford Um die Proteinkonzentration bestimmen zu können, musste zunächst eine Protein-Standard Reihe mit den Konzentrationen 0,7 µg/ml, 4,2 µg/ml, 9,8 µg/ml, 14 µg/ml, 21 µg/ml und 42 µg/ml pipettiert werden. Die zu messenden Proben wurden wie folgt pipettiert: zu 796 µl H2O wurden jeweils 4 µl Probe und 200 µl Bradford Reagenz dazugegeben und gut gemischt. Ein Farbumschlag nach blau zeigte an, ob Proteine enthalten waren. Die Ansätze wurden in Plastikküvetten überführt und die Konzentrationsbestimmung fand an einem Photometer statt. Die angezeigte Konzentration wurde in die Formel Konzentration x 250 (Verdünnung) : 1000 = X µg/µl eingesetzt um die Einheit µg/µl zu erhalten SDS-Polyacrylamidgelelektrophorese (SDS-PAGE) Durch SDS-Polyacrylamid-Gelelektrophorese lassen sich Proteingemische nach ihrem Molekulargewicht auftrennen. Die folgende Methode wurde nach Laemmli durchgeführt (Laemmli, 1970). Es wurden je nach Molekulargewicht der aufzutrennenden Proteine Polyacrylamidtrenngele mit einer Acrylamidkonzentration zwischen 5-15% angefertigt (siehe Tabelle 2.1). Auf das auspolymerisierte Trenngel wurde anschließend ein Sammelgel mit 5,6%iger Acrylamidkonzentration gegossen. Pro Tasche wurden 18 µl aufgetragen, die sich aus µg Protein, 1x Ladepuffer und H 2 O zusammensetzten. Danach erfolgte eine Denaturierung bei 95 C für 5 min auf einem Heizblock. Die Proben wurden gemeinsam mit einem Molekulargewichtsstandard (BioRad) auf das Gel geladen. Die SDS-Gelelektrophorese wurde in einer Mini Protean 3 Kammer (BioRad) mit 1x Laufpuffer bei V für min durchgeführt Transfer von Proteinen auf PVDF-Membranen Nach dem Auftrennen eines Proteingemisches durch die SDS-Polyacrylamidgelelektrophorese wurden die Proteine elektrophoretisch aus dem Polyacrylamidgel auf eine PVDF- Membran transferiert. Der Transfer erfolgte nach dem Nass-Blot-Verfahren in 1x Transfer Puffer. Folgender Blot-Aufbau wurde dafür gewählt: Auf die Anode wurde ein in Transferpuffer getränkter Schwamm und 1 Whatmanpapier gelegt, auf das das Gel platziert wurde. Auf das Gel wurde die PVDF-Membran, die zuvor 1 min in Methanol, 1 min in H 2 0 und 1 min in 1x Transferpuffer getränkt wurde, gestapelt, auf die wiederum 1 Whatmanpapier und ein Schwamm gelegt wurde. Der Transfer erfolgte für 1,5-4 h bei 0,6 ma/sq. cm. 39
47 2 Material und Methoden Immunblotanalyse und Detektion durch verstärkte Chemilumineszenz (ECL) Nach dem Transfer wurde die PVDF-Membran über Nacht in eine 3-5%ige TBST-Milch Blockierlösung gegeben, um unspezifische Bindungen der eingesetzten Antikörper an die Membran zu verhindern. Die anschließende Inkubation mit dem Primärantikörper erfolgte je nach Antikörper in TBST oder 3%-TBST-Milch für 1-4 h bei RT oder über Nacht bei 4 C auf einem Kippschüttler. Danach wurde die Membran 3x 15 min auf einer Wippe in TBST gewaschen. Die Inkubation mit dem sekundären Antikörper 1:2000-1:5000, verdünnt in TBST oder 3% TBST-Milch, erfolgte 1 h bei RT. Gewaschen wurde wieder 3x 15 min mit TBST. Für die anschließende Immundetektion wurde das ECL (Enhanced Chemi- Luminescent) Plus Western Blot Detektions Kit (GE Healthcare) verwendet. Die Membran wurde mit 2 ml ECL Reagenz und 50 µl Substrat bedeckt und für 5 min inkubiert. Die Chemilumineszenz wurde mittels Röntgenfilm ermittelt Indirekte Immunfluoreszenz ALT-assoziierte PML-Bodies (APBs) Bei ALT-assoziierten PML-Bodies kommt es zu einer Kolokalisation von dem Telomerbindeprotein TRF2, Promyelozytischen Leukämie Kernkörperchen (PML-Bodies), Telomer-DNA und Proteinen die bei der homologen Rekombination eine Rolle spielen, wie z.b. Rad52. Als indirekten Nachweis für ALT wurden Färbungen gegen TRF2 und PML durchgeführt und nach Kolokalisationen untersucht. Dazu wurden die zu untersuchenden Zelltypen in 6-Loch Platten auf Deckgläser mit dem Durchmesser von 25 mm ausplattiert und für 1-2 Tage im Brutschrank bei 37 C und 5% CO 2 inkubiert. Dann wurden die mit Zellen bewachsenen Deckgläser in neue 6-Loch Platten überführt, zweimal mit PBS gewaschen und mit 3,7% Formaldehyd in PBS für 10 min bei RT auf dem Schüttler fixiert. Nach weiteren zwei Waschschritten mit PBS wurden die Zellen mit 0,5%igem Nonidet P40 verdünnt in PBS permeabilisiert. Dann wurden die Zellen einmal mit PBS gewaschen und anschließend 1 h oder über Nacht mit PBG (0,5%iges BSA, 0,1%iges Cold Water Fish Gelatin in PBS) blockiert. Auf diesen Schritt folgend, wurden zunächst pro Deckglas 100 µl TRF2-Antikörper (1:600) zugegeben und über Nacht bei 4 C, eingewickelt in feuchte Tücher, inkubiert. Am nächsten Tag wurden die Zellen 3x mit PBS gewaschen und dann je 100 µl des PML- Antikörpers (1:10000) dazugegeben und 1-4 h bei RT inkubiert. Es folgten wieder drei Waschschritte für je 5 min mit PBS. Als Zweitantikörper wurden ein Cy3-gekoppelter anti- Ziege 1:500 (Jackson ImmunoResearch) und ein Alexa Fluor 594 anti-maus 1:500 (Molecular Probes) Antikörper eingesetzt und 1 h lichtgeschützt inkubiert. Letztlich wurde 3x 5 min mit PBS gewaschen und die Deckgläser mit der Zellseite nach unten auf einen mit einem Tropfen Slowfade Reagenz benetzten Objektträger gelegt und mit Nagellack fixiert. Die Auswertung wurde an einem konfokalen Mikroskop der Firma Leica (Leica TCS SP2 40
48 2 Material und Methoden AOBS) durchgeführt. Es wurde ein 63faches Objektiv verwendet und jeweils zwischen 4 und 14 Ebenen fotografiert. Die Kolokalisationen von TRF2 und PML wurden ausgezählt und in einer Excel-Tabelle dokumentiert (0 APBs, 1 APB, 2 APBs, 3 APBs und mehr pro Zelle) Fluoreszenz in situ Hybridisierung (FISH) Quantitative Fluoreszenz in situ Hybridisierung (Q-FISH) Bei der quantitativen Fluoreszenz in situ Hybridisierung werden durch Metaphasenpräparation ausgebreitete Chromosomen mit einer PNA-Telomer Sonde, die mit dem Fluoreszenzfarbstoff Cy3 markiert ist, hybridisiert. Nach Gegenfärbung der Chromosomen mit DAPI können diese unter dem Fluoreszenzmikroskop dargestellt und die Fluoreszenzintensitäten der Telomere gemessen werden. Der Vorteil bei dieser Methode ist, dass man Telomermessungen an Einzelzellen durchführen kann. Die Q-FISH Methode wurde, wie von Lansdrop et al beschrieben, mit geringen Modifikationen durchgeführt (Lansdorp et al., 1996). Zur Metaphasenpräparation wurden zunächst die unterschiedlichen Zellen in 10 cm Zellkulturplatten ausgesät und im Brutschrank wachsen gelassen, bis sie eine Konfluenz zwischen 50-70% erreicht hatten. Um die Zellen dann in der Metaphase zu arretieren, wurden sie mit einer Colcemid-Konzentration (Invitrogen) von 0,1 µg/ml in Medium für 6 h behandelt. Anschließend wurden die Zellen abtrypsiniert, für 5 min bei 138 g abzentrifugiert, das Pellet in 2 ml 75 mm KCl vorsichtig resuspendiert und für min bei 37 C inkubiert. Hierbei kam es zur erwünschten hypotonen Schwellung der Zellen. Zur Fixierung wurden zunächst 10 Tropfen Methanol/Eisessig (3:1) dazugegeben und dann 5 min bei 138 g zentrifugiert. Der Überstand wurde verworfen und das Pellet wurde in 2 ml Methanol/Eisessig resuspendiert und 5 min bei 138 g abzentrifugiert. Dieser Schritt wurde 2-3x wiederholt. Anschließend wurden die Zellen entweder bei -20 C gelagert oder aus einer Entfernung von cm auf einen Objektträger getropft. Bei dieser Handhabung sollten die Zellen platzen, und wenn sich eine Zelle in der Metaphase befindet die Chromosomen gut verteilt werden. Ob sich genug Metaphasen auf dem Objektträger befanden, wurde am Durchlichtmikroskop überprüft. Zur Vorbereitung der Hybridisierung wurden die Objektträger mit den Metaphasen 15 min in 1x PBS (Invitrogen) rehydriert, 2 min mit 3,7% in PBS verdünntem Formaldehyd fixiert und noch mal 3x 5 min mit PBS gewaschen. Zum Verdau von Proteinanteilen, die das Binden der Hybridisierungssonde an die Telomere behindern könnten, wurden die Objektträger 20 min im Wasserbad bei 37 C in einer wässrigen Pepsinlösung (550 U/ml, ph2) inkubiert, anschließend einmal 2 min mit PBS gespült und nochmals 2 min in 3,7% Formaldehyd fixiert. Nach dreimaligem Waschen mit PBS wurden die Zellen mit einer aufsteigenden Alkoholreihe (70%, 90% und 100%) je 5 min dehydriert und anschließend luftgetrocknet. Als nächstes wurden 2x 10 µl des Hybridisierungsmixes auf ein Deckglas 41

49 2 Material und Methoden pipettiert und der Objektträger vorsichtig und unter Vermeidung von Luftblasen mit den Zellen nach unten auf das Deckglas platziert. Zur DNA-Denaturierung wurden die Zellen für 3 min auf einen 80 C heißen Heizblock gelegt und anschließend für 2-12 h in einer dunklen, feuchten Kammer hybridisiert. Auf die Hybridisierung folgten zwei Waschschritte je 15 min mit Waschlösung I (70% Formamid, 0,1% BSA, 10 mm Tris-HCl, ph7-7,5) und 3x je 5 min mit Waschlösung II (0,1 M Tris-HCl, 0,15 M NaCl, 0,08% Tween-20). Letztlich wurden die Zellen in einer aufsteigenden Alkoholreihe 70%, 90% und 100% je 5 min dehydriert und luftgetrocknet. Zum Schutz des Fluoreszenzfarbstoffes vor dem Ausbleichen und zum Anfärben von Chromosomen, wurden auf ein frisches Deckglas 2 Tropfen Vectashield +DAPI gegeben, dieses vorsichtig auf den mit den Zellen enthaltenen Objektträger gelegt und mit Nagellack fixiert. Die hybridisierten und angefärbten Metaphasen wurden am Fluoreszenzmikroskop betrachtet und mit Hilfe einer digitalen Kamera aufgenommen. Durch verschiedene vorgeschaltete Filter konnte man entweder die DAPI-angefärbten (395 nm Filter) ganzen Chromosomen oder die Cy-3 (560 nm Filter) markierten Telomere darstellen (siehe Abbildung 2.3) Abb. 2.3 Metaphasen nach Fluoreszenz in situ Hybridisierung. Durch Vorschalten spezieller Filter können entweder die DAPI angefärbten Chromosomen oder die mit Cy-3 markierten Telomere dargestellt werden (in diesem Bild übereinander gelagert). Diese Aufteilung ist notwendig, um zunächst über den DAPI-Filter geeignete gut ausgebreitete Chromosomen ausfindig zu machen und nachfolgend über den Cy-3-Filter die Telomere zu erfassen. Die Bilder wurden als Bitmap gespeichert. Die Auswertung der so erhaltenen Aufnahmen erfolgte durch zwei verschiedene Untersucher, indem die Signale als heterogen oder homogen bewertet wurden. Die Werte wurden in einer Excel-Tabelle aufgelistet in Prozent umgerechnet und davon ein Diagramm erstellt. In der vorliegenden Arbeit wurden die Q-FISH-Analysen an mindestens Metaphasen einer Zellpopulation pro Versuch durchgeführt Chromosomen Orientierungs- Fluoreszenz in situ Hybridisierung (CO-FISH) Bei dieser Methode kann der Austausch von Telomeren an Schwesterchromatiden und Chromosomen an Einzelzellen demonstriert werden (Abbildung 2.4). 42
50 2 Material und Methoden Die zu untersuchenden Zellen wurden trypsiniert und so ausplattiert, dass sie am nächsten Tag eine Konfluenz zwischen 40-60% erlangten. Das jeweils zellspezifische Medium wurde mit einem Verhältnis von 3:1 5 BrdU:5 BrdC mit einer Endkonzentration von 1x10-5 M versetzt. Die Zellen wurden h im Brutschrank bei 37 C inkubiert und vor Lichteinfluss geschützt. Bei diesem Schritt werden während der S-Phase die modifizierten BrdU und BrdC-Nukleotide in den neu synthetisierten DNA-Strang eingebaut. Nach h wurde zu den Zellen, um sie in der Metaphase zu arretieren, eine Colcemid-Konzentration (Invitrogen) von 0,1 µg/ml für 6 h ins Medium dazugegeben. Anschließend wurden die Zellen abtrypsiniert und abzentrifugiert (138 g, 4 C) und das Pellet in 2 ml vorgewärmtes 75 mm KCl vorsichtig resuspendiert. Es folgte eine lichtgeschützte Inkubation von min im Wasserbad bei 37%. Nach dieser Inkubationszeit wurden vorsichtig 5 Tropfen 3:1 eisgekühlter Methanol/Eisessig zugegeben und 5 min bei 138 g zentrifugiert. Dann wurde das Pellet in 2 ml 3:1 Methanol/Eisessig resuspendiert und anschließend wieder bei 138 g 5 min zentrifugiert. Dieser Schritt wurde 3-4x wiederholt und das Pellet dabei im Dunkeln gehalten. Als nächstes wurden die Zellen in Methanol/Eisessig entweder bei -20 C gelagert oder direkt aus einer Entfernung von 20 cm auf die Objektträger getropft, damit die Zellen die Gelegenheit haben, nach dem Anschwellen zu platzen. Die Objektträger wurden für eine Minute auf einen mit feuchten Tüchern bedeckten Heizblock bei 42 C gelegt und anschließend über Nacht trockenen gelassen. Die Objektträger wurden 5 min in PBS auf einer Wippe rehydriert und anschließend mit 0,5 mg/ml RNaseA in PBS für 10 min bei 37 C inkubiert, um RNAs von den Telomeren zu befreien. Im nächsten Schritt wurden die Objektträger in eine 0,5 µg/ml Hoechst 33258/2xSSC-Lösung gegeben, um die DNA für den nachfolgenden Schritt, eine UV-Behandlung, sensitiver zu machen. Dafür wurden die Objektträger in ein Plastikgefäß gelegt und mit 2x SSC bedeckt und für 30 min mit einer Wellenlänge von 365 nm (oder der Energie von 5.4 x103 J/m2) im einem Stratalinker 2400 UV irradiator (Artisan Scientific) bestrahlt, was zu Strangbrüchen an den BrdU und BrdC modifizierten Basen führte. Anschließend wurde der neusynthetisierte und aufgebrochene Strang mit 3 U/µl Exonuclease III in dem dazugehörigen Puffer 10 min bei RT verdaut. Übrig blieb nur der parentale Strang. Dann wurden die Objektträger 5 min in PBS gegeben und in einer aufsteigenden Alkoholreihe (70%, 90% und 100%) dehydriert. Die Objektträger wurden entweder bei RT gelagert oder es wurde mit der Hybridisierung weitergemacht. Für die Hybridisierung wurde zunächst der Hybridisierungsmix mit einer Telomersonde (Tel-FITC- PNA-Sonde) vorbereitet. Es wurden je 20 µl des Hybridisierungsmixes auf ein Deckglas pipettiert und der Objektträger vorsichtig und unter Vermeidung von Luftblasen mit den Zellen nach unten auf das Deckglas platziert. Die Hybridisierung erfolgte über Nacht in einer lichtgeschützten, feuchten Kammer bei RT. Nach dieser Hybridisierung erfolgte die Hybridisierung mit der zweiten (Tel-Cy3-PNA-Sonde) Sonde in der gleichen Zusammen- 43
")
51 2 Material und Methoden setzung wie die FITC-Sonde aber für nur 2-3 h. Danach wurden die Objektträger 2 mal 15 min mit Waschlösung 1 gewaschen, dann 3 mal 5 min mit Waschlösung 2 gewaschen und schließlich in einer aufsteigenden Ethanolreihe (70%, 90% und 100%) dehydriert. Nach Trocknen der Objektträger wurden je 2 Tropfen DAPI auf ein Deckglas gegeben und die Objektträger unter Vermeidung von Luftblasen mit den Zellen nach unten auf das Deckglas gegeben. Die Deckgläser wurden mit Nagelack fixiert und die Objektträger bei 4 C aufbewahrt. Bilder wurden mit einem Fluoreszenzmikroskop der Firma Zeiss mit einer 63 fachen Vergrößerung gemacht. Rekombinationsereignisse, also eine Kolokalisation beider Sonden wurden von Hand ausgezählt. Normale Replikation Replikation und Telomer- Schwesterchromatidaustausch Replikation in BrdU und BrdC Telomer-Schwesterchromatidaustausch Verdau des neusynthetisierten Strangs 1 Probe 2 Proben 1 Probe 2 Proben Abb. 2.4 Schematische Darstellung des Chromosomen-Orientierungs-FISH Bei der DNA-Replikation kommt es beim neusynthetisierten Strang zum Einbau von BrdU und BrdC. Im nächsten Schritt wird der neusynthetisierte Strang durch die Exonuclease III abgebaut. Kam es vorher zu einer Rekombination zwischen Schwesterchromatiden, kommt es zu einer Kolokalisation der Cy3-markierten C-reichen Sonde mit der FITC-markierten G-reichen Sonde. Abbildung aus (Bechter et al., 2004) Fluoreszenz-aktivierendes Zellsortieren (FACS) FACS-Analysen können neben vielen anderen Anwendungen auch zur Bestimmung des Zellzyklus verwendet werden. Da die Zellen in der G1-Phase nur über einen einfachen Chromosomensatz verfügen, lassen sich diese Zellen bei einer bestimmten Intensität durch die mit der DNA interagierenden und fluoreszierenden Substanz Propidiumiodid detektieren. Zellen in der G2-Phase besitzen einen doppelten Chromosomensatz, was zu einer Zellpopulation führt, die sich bei der doppelten Intensität an fluoreszierendem Propidiumiodid detektieren lässt. Neben einem haploiden oder diploiden Chromosomensatz kann man auch einen aneuploiden Chromosomensatz detektieren. 44
52 2 Material und Methoden Um das Ploidiestadium der CP-A-Zellen zu bestimmen, wurden die Zellen mit Trypsin von einer 10 cm Kulturplatte gelöst, in PBS aufgenommen und gezählt. Je Ansatz wurden mindestens 1x Zellen in 0,5 ml PBS aufgenommen und anschließend bei 200 g 6 min zentrifugiert. Das Zellpellet wurde in 4,5 ml 70%igen Ethanol aufgenommen und die Zellen mindestens 2 h auf Eis fixiert. Auf diesen Schritt folgend, wurden die Zellen 10 min bei 400 g zentrifugiert, das Ethanol entfernt und in 5 ml PBS resuspendiert und erneut bei 400 g zentrifugiert. Dieser Schritt wurde nochmals wiederholt, anschießend die Zellen in 1 ml PBS resuspendiert und 25 µl Propidiumiodid (1 mg/ml) hinzugefügt. Die Zellen waren von nun an vor Lichteinstrahlung zu schützen. Die Messung erfolgte an einem FACSCalibur der Firma Becton Dickinson. In der Auswertung wurde die gemessene Propidiumiodidintensität von einer definierten Anzahl von Zellen in einem Diagramm graphisch dargestellt. In Dieser Darstellung zeigt sich ein bestimmtes Verteilungsmuster der Zellen, die dann den einzelnen Phasen des Zellzyklus zugeordnet werden konnte. 45

53 3 Ergebnisse 3 Ergebnisse 3.1 Charakterisierung der verschiedenen Zelllinien Charakterisierung des Barrettzell-Modells Die Barrettzelllinie CP-A wurde aus chirurgischen Biopsaten von Patienten mit Barrett- Ösophagus gewonnen, in Kultur genommen und als eine Barrettzelllinie, die eine frühe Stufe der Barrett-Adenokarzinogenese repräsentiert, beschrieben (Palanca-Wessels et al., 1998). Um zu überprüfen, ob diese Barrettzelllinie wirklich eine frühe Stufe der Barrett- Adenokarzinogenese widerspiegelt, wurde diese auf p53 und p16 mittels Western Blot- Analyse überprüft. Wie in Abbildung 3.1 A dargestellt, sind die CP-A-Zellen p53 intakt. Es ist nur ein schwaches Signal bei einer Größe von 53 kda sichtbar, da dies Zellzyklus-abhängig reguliert wird und somit schnell in der Zelle durch Ubiquitinierung abgebaut wird. In den p53- defizienten CP-B-Zellen erscheint eine dicke Bande auf der Höhe von 53 kda, da sich das Tetramer p53, wenn auch nur ein Allel mutiert ist, in der Zelle akkumuliert. Dieses kann jedoch seine Aufgaben in der Zelle nicht mehr erfüllen. A B p53 p16 ß-Tubulin ß-Tubulin CP-A-Zellen 2 CP-B-Zellen CP-A-Zellen 2 CP-C-Zellen Abb. 3.1: p53- und p16-western Blot in der Barrettzelllinie CP-A Nach Ernte der CP-A-Zellen wurden die Proteine mit CHAPS-Lysepuffer extrahiert und 75 µg Protein durch eine SDS-Polyacrylamidgelelektrophorese aufgetrennt und auf eine PVDF-Membran transferiert. Für die Immunblotanalyse wurden, als primäre Antikörper, ein anti-p53-antikörper (A) und ein anti-p16-antikörper (B) verwendet. Als Ladekontrolle wurde anti-ß-tubulin eingesetzt. Als p53 negativ Kontrolle wurde die Barrettzelllinie CP-B verwendet und als p16 positiv Kontrolle die Barrettzelllinie CP-C. Wie im p16-western Blot in Abbildung 3.1 B sichtbar, sind die CP-A-Zellen p16 defizient. Die CP-C-Zelllinie dagegen zeigt ein intaktes p16-protein. Im nächsten Schritt sollte überprüft werden, ob die p16-defizienz in den CP-A-Zellen durch die Methylierung des p16-promotors verursacht wird, da dies in vielen Dysplasien der Barrettkarzinogenese der Fall ist (Morales et al., 2002). Hierfür wurde eine methylierungsspezifische PCR durchgeführt. Wie in Reihe 2, Abbildung 3.2 dargestellt, ist der p16-promotor der CP-A-Zellen nicht methyliert, wie auch der Promotor der CP-C-Zellen, in denen das p16 Protein im Western Blot nachgewiesen 46

54 3 Ergebnisse werden konnte. Im Falle eines unmethylierten Promotors und spezifischen Primern erscheint jeweils ein positives DNA-Signal auf der Höhe von 151 bp, dagegen bei der artifiziell methylierten Positivkontrolle und hierfür spezifischen Primern ein DNA-Signal bei 150 bp. Dies zeigt, dass die Inaktivierung von p16 in den CP-A-Zellen nicht an dem Methylierungsstatus des p16 Promotors liegt. 400 bp 300 bp 200 bp 151 bp 150 bp 100 bp 1 CPA-Zellen: Oligonukleotide für methylierten p16 Promotor 2 CPA-Zellen: Oligonukleotide für unmethylierten p16 Promotor 3 CPC-Zellen: Oligonukleotide für methylierten p16 Promotor 4 CPC-Zellen: Oligonukleotide für unmethylierten p16 Promotor 5 pos. Kontrolle: Oligonukleotide für methylierten p16 Promotor 6 pos. Kontrolle: Oligonukleotide für unmetylierten p16 Promotor Abb. 3.2 Methylierungsspezifische PCR des p16-promotors in CP-A- und CP-C-Zellen Auf die DNA-Extraktion aus den CP-A- und CP-C-Zellen folgte eine Bisulfitbehandlung von 2 µg DNA. Im nächsten Schritt wurde eine PCR mit spezifischen Primern für den Promotormethylierungsstatus (p16mforward, p16mreverse) und für den unmethylierten Status (p16forward, p16reverse) durchgeführt. Die PCR-Produkte wurden auf einem 6%igen Acrylamidgel aufgetrennt und unter einem UV- Spektrometer betrachtet und fotografiert. Als Negativkontrolle wurde anstelle von DNA Wasser eingesetzt und als Positivkontrolle wurde eine artifiziell methylierte DNA verwendet. Als nächstes wurde der DNA-Gehalt bzw. das Ploidiestadium der Zellen mit Hilfe einer FACS-Analyse untersucht. In ganz frühen Stadien der Barrett-Adenokarzinogenese wurde beschrieben, dass Barrettzellen einen diploiden Chromosomensatz besitzen. Ein wichtiger Schritt auf dem Weg zum Barrett-Adenokarzinom ist das Erlangen eines polyploiden bzw. aneuploiden Chromosomensatzes. Die CP-A-Zellen besitzen, wie in Abbildung 3.3 dargestellt einen diploiden Chromosomensatz, mit Zellen in der G1-, S- und G2-Phase. 47
55 3 Ergebnisse 200 Zellzahl Propidiumiodid 1000 Abb. 3.3: FACS-Analyse zur Bestimmung des Ploidiestadiums der CP-A-Zellen Es wurden 1x Zellen in der FACS-Analyse untersucht, die zunächst 2 h in 70% Ethanol fixiert, dann mit PBS gewaschen und schließlich mit Propidiumiodid gefärbt wurden. Die Durchführung der FACS-Analyse erfolgte an einem FACSCalibur Gerät. 1: G1-Phase; 2: S-Phase; 3: G2-Phase Weiter wurde das Wachstumsverhalten der CP-A-Zellen untersucht. Dafür wurden 1x 10 5 Zellen ausplattiert und 3 Tage später gezählt und mit Hilfe einer Formel die Populationsdopplungen bestimmt. Es wurde berechnet, dass sich die CP-A-Zellen im Schnitt einmal am Tag teilen. Zusammenfassend lässt sich sagen, dass die Barrettzelllinie CP-A eine sehr frühe Stufe der Barrett-Adenokarzinogenese darstellt: Die Zellen weisen eine p16-defizienz auf und sind mit htert immortalisiert. Das Tumorsuppressorgen p53 ist jedoch intakt und die Zellen besitzen einen diploiden, nicht polyploiden oder aneuploiden Chromosomensatz. Somit kann die Barrettzelllinie CP-A als eine genetisch definierte, prämaligne Stufe der Barrett- Adenokarzinogenese betrachtet werden. Eine Tumorzelllinie auf der Seite der Barrett-Adenokarzinogenese ist die kommerziell erworbene Adenokarzinomzelllinie OE19. Hier waren die genetischen Alterationen nicht genau bekannt, nur dass diese Zellen aus einem Tumor, der als pathologisches Stadium III eingestuft wurde, stammen. Diese repräsentiert somit eine genetisch nicht definierte Tumorzelllinie Charakterisierung des zellulären Plattenepithelkarzinogenese-Modells Eine prämaligne Stufe der Plattenepithelkarzinogenese stellen immortale epitheliale Keratinozyten, die als EPC-hTERT bezeichnet werden, dar. Die EPC-Zellen wurden aus Biopsien von gesunden Spendern in Kultur genommen und anschließend mit htert immortalisiert (Quante et al., 2005). Diese Zellen dienen als Kontrollzellen, da bei ihnen bereits ein Wechsel von Telomerase zu ALT gezeigt werden konnte (Thaler, 2009). Eine spätere Stufe der Plattenepithelkarzinogenese repräsentiert eine genetisch definierte Plattenepithelkarzinomzelllinie, die OKF6 D1/dnp53/EGFR/myc-Zellen. Die OKF6-Zellen 48

56 3 Ergebnisse wurden ebenfalls einem gesunden Spender entnommen und in Kultur genommen. Diese wurden anschließend schrittweise durch Überexpression von Cyclin D1, dominant-negativem p53, EGFR und c-myc maligne transformiert. Dabei rekrutierte diese Zelllinie bereits nach Überexpression von Cyclin D1 und dnp53 den ALT-Mechanismus zur Immortalisierung, wobei in der weiteren Tumorprogression nach Überexpression von EGFR die Telomerase aktiviert wurde Charakterisierung der Telomeraseaktivität und der Telomerlängen in der genetisch definierten ösophagealen Plattenepithelkarzinomzelllinie, der genetisch definierten immortalen Barrettzelllinie und der genetisch nicht definierten Adenokarzinomzelllinie Alle drei Zelllinien wurden zunächst auf einen stabilen Telomerasegehalt mittels Western Blot auf Proteinebene und im TRAP-Assay auf Telomeraseaktivität untersucht. Wenn sich eine stabile Telomeraseaktivität nachweisen lässt, sollten im nächsten Schritt in den verschiedenen Zelllinien die Telomerlängen mit Hilfe von TRF-Analysen mit anschließendem Southern Blot gemessen und verglichen werden. Dafür wurden die genetisch definierten, ösophagealen Plattenepithelkarzinomzellen (OKF6 D1/dnp53/EGFR/myc), die genetisch definierten, immortalisierten Barrettzellen (CP-A) und die genetisch nicht definierten Adenokarzinomzellen (OE19) auf ihren Telomerasegehalt zunächst in einem Western Blot untersucht. In Abbildung 3.4 ist der Western Blot gegen die enzymatische Untereinheit der Telomerase htert dargestellt. In allen drei Zelllinien konnte das Protein bei einer Größe von 220 kda nachgewiesen werden. ß-Tubulin wurde als Ladekontrolle mitgeladen (Abbildung 3.4). 220 kda htert 57 kda ß-Tubulin OKF6 D1/p53/EGFR/myc 2 CP-A 3 OE19 Abb. 3.4: htert-western Blot in OKF6 D1/dnp53/EGFR/myc-, CP-A- und OE19-Zellen Die Proteinextraktion aus OKF6 D1/p53/EGFR/myc-, CPA- und OE19-Zellen erfolgte mit CHAPS- Lysepuffer. Es wurden je 75 µg Protein auf einem 6%igen Acrylamidgel aufgetrennt und anschließend auf eine PVDF-Membran transferiert. Zur Immunblotanalyse wurden als primäre Antikörper ein antihtert-antikörper und zur Ladekontrolle ein anti-ß-tubulin Antikörper eingesetzt. Die Chemilumineszenz wurde auf einem Röntgenfilm detektiert. 49

57 3 Ergebnisse Da die Telomerase im Western Blot auf Proteinebene nachgewiesen werden konnte, sollte als nächstes die funktionelle Aktivität der Telomerase in allen drei Zelllinien untersucht werden. Um dies zu überprüfen wurde ein TRAP-Assay durchgeführt. Dieser ist eine sehr sensitive und somit die gängigeste Methode um eine Telomeraseaktivität nachzuweisen. In allen drei Zelllinien konnte, wie in Abbildung 3.5 A sichtbar, eine funktionell aktive Telomerase nachgewiesen werden, die in der Lage war ein radioaktiv-markiertes Telomerähnliches Oligonukleotid zu verlängern. Die Aktivität korreliert mit Stärke und Höhe der Bandenleiter. HI- A B 21 kbp 5 kbp 1 OKF6 D1/p53/EGFR/myc-Zellen 2 CP-A-Zellen 3 OE19-Zellen IK Abb. 3.5: Nachweis der Telomeraseaktivität und der Telomerlängen in den OKF6 D1/dnp53/EGFR/myc-, CP-A- und OE19-Zellen (A) Nach Ernte der OKF6 D1/dnp53/EGFR/myc-, CP-A- und OE19-Zellen wurden diese mit CHAPS- Lysepuffer lysiert und 30 ng an Protein für den TRAP-Assay eingesetzt. Die radioaktiven PCR- Produkte wurden auf einem Acrylamidgel aufgetrennt, das Gel getrocknet und das Signal auf einem Röntgenfilm detektiert. HI: Hitzeinaktiviert; IK: interne Kontrolle (B) Aus den OKF6 D1/dnp53/ EGFR/myc-, CP-A- und OE19-Zellen wurde die genomische DNA isoliert und anschließend 5 µg mit den Enzymen HinfI und RsaI verdaut. Der Verdau wurde auf einem 0,5%igen Agarosegel aufgetrennt, auf eine Hybond-XL Membran transferiert und die Telomere mit einer radioaktiv-markierten Sonde gegen die Telomersequenz auf einem Röntgenfilm sichtbar gemacht. Anhand einer TRF-Analyse mit folgender Southern Blot-Analyse wurden die Telomerlängen in den drei Zelllinien untersucht und verglichen (Abbildung 3.5 B). Die Telomerlänge in den genetisch definierten Plattenepithelkarzinomzellen lag bei einer Durchschnittslänge von ca. 19 kbp. Die recht scharf abgetrennte Bande lässt auf homogene Telomere aufgrund stabiler Telomeraseaktivität in einer Zellpopulation schließen. Die genetisch definierte Barrettzelllinie dagegen hatte nur eine Durchschnittstelomerlänge von ca. 12 kbp. Die Telomerlänge ist aber ebenso wie bei den genetisch definierten Plattenepithelkarzinomzellen sehr homogen, da die Bande scharf begrenzt ist. Die Telomerlänge der Adenokarzinomzelllinie ist im Vergleich zu 50
58 3 Ergebnisse den genetisch definierten Plattenepithelkarzinomzellen und den genetisch definierten Barrett- Zellen kürzer und lag zwischen 7 kbp und 12 kbp. Im Vergleich zu den anderen beiden Zelllinien ist die Bande im Southern Blot jedoch nicht scharf abgetrennt, und die Telomerlängen in der Adenokarzinomzelllinie damit deutlich heterogener. Die Telomeraseaktivität, sowie die Telomerlängen änderten sich nicht bei Betrachtung unterschiedlicher Passagen. 3.2 Genetische Inhibition der Telomerase führt zur Induktion eines ALT-Mechanismus in genetisch definierten Zelllinien und in der genetisch nicht definierten Adenokarzinomzellen In vorangegangenen Untersuchungen konnte gezeigt werden, dass htert-immortalisierte ösophageale Keratinozyten im Zellzyklusarrest den Telomererhaltungsmechanismus von Telomerase zum ALT-Mechanismus wechseln können (Von Werder, 2007). Weiter konnte in unserer Arbeitsgruppe demonstriert werden, dass in diesen htert-immortalisierten ösophagealen Keratinozyten die Telomerase genetisch inhibiert werden kann und in TRF- Analysen die Telomere lang blieben oder sogar verlängert wurden (Thaler, 2009). Um dies zu bestätigen und die Stabilität dieses Mechanismus darzustellen, wurden die Telomeraseinhibierten und Telomerase-positiven (Kontrollzellen), immortalisierten ösophagealen Keratinozyten auf ihren Telomerasegehalt, sowie auf ihre Telomerlängen über die Zeit untersucht (Abb. 3.6). Wie in Abbildung 3.6 dargestellt, wird die Telomeraseaktivität durch Transduktion mit den Telomeraseinhibitoren Plasmid 3 und 6 (Tab. 1) stark herrunter reguliert. Auch die Telomerlängen bleiben nach Transduktion mit Plasmid 6 lang, verglichen mit den Kontrollzellen, die Plasmid 1 enthalten. Auch nach zahlreichen Passagen zeigen diese ALT-spezifische Eigenschaften, wie heterogene Telomerlängen. 51
 Nach Ernte der EPC-hTERT-Zellen wurden diese mit CHAPS-Lysepuffer lysiert und 30 ng an Protein für den TRAP-Assay eingesetzt.")
 Aus den EPC-hTERT-Zellen wurde die DNA isoliert und anschließend 5 µg mit HinfI und RsaI verdaut.")
59 3 Ergebnisse HI- A B 21 kbp IK- 5 kbp EPC-hTERT-Zellen Plasmid 1 2 EPC-hTERT-Zellen Plasmid 3 3 EPC-hTERT-Zellen Plasmid 6 Abb. 3.6: Nachweis der Telomeraseaktivität und der Telomerlängen nach Telomeraseinhibierung in EPC-hTERT-Zellen. (A) Nach Ernte der EPC-hTERT-Zellen wurden diese mit CHAPS-Lysepuffer lysiert und 30 ng an Protein für den TRAP-Assay eingesetzt. Die amplifizierten radioaktiv-markierten PCR-Produkte wurden auf einem Acrylamidgel aufgetrennt, das Gel getrocknet und das Signal auf einem Röntgenfilm über Nacht bei -80 C detektiert. HI: Hitzeinaktiviert; IK: interne Kontrolle. (B) Aus den EPC-hTERT-Zellen wurde die DNA isoliert und anschließend 5 µg mit HinfI und RsaI verdaut. Der Verdau wurde auf einem 0,5%igen Agarosegel aufgetrennt, auf ein Hybond-XL Membran transferiert und die Telomere mit einer radioaktiv-markierten Sonde komplementär zur Telomersequenz sichtbar gemacht, indem das Signal auf einem Film detektiert wurde. Der nächste Schritt war nun, die Telomerase in den genetisch definierten Plattenepithelkarzinomzellen (OKF6 D1/dnp53/EGFR/myc) und Barrettzellen (CP-A) sowie in den genetisch nicht definierten Adenokarzinomzellen (OE19), so wie für die immortalisierten, ösophagealen Keratinozyten etabliert und optimiert, gezielt über genetische Inhibition auszuschalten. Durch Untersuchungen an den immortalisierten ösophagealen Keratinozyten, stand fest, welche Plasmide am besten für eine Telomeraseinhibition geeignet sind. Diese wurden daraufhin in den drei Zelltypen eingesetzt (Tab. 1). So sollte untersucht werden, ob auch in späteren Stufen der Karzinogenese sowie unterschiedlichen Tumorentitäten bzw. Differenzierungen durch Inhibition der Telomerase ein ALT-Mechanismus induziert werden kann. Nach eingehender Charakterisierung der Telomeraseaktivität und der Telomerlängen in den genetisch definierten Plattenepithelkarzinomzellen, den genetisch definierten Barrettzellen und den nicht definierten Adenokarzinomzellen wurden diese nun mit folgenden Telomeraseinhibitoren lentiviral transduziert (Li et al., 2004): 52
60 3 Ergebnisse Tabelle 1: Auflistung der verwendeten Telomeraseinhibitoren und Kontrollplasmiden Plasmid Nummer Bezeichnung Funktion 1 WT-hTER Wildtyp RNA-Matrize 2 MT-hTER/AU5 Mutierte RNA-Matrize 3 MT-hTER/47/A Mutierte RNA-Matrize 6 MT-hTER/AU5+siRNA Mutierte RNA-Matrize in Kombination mit einer sirna gegen die RNA- Matrize 7 MT-hTER/47A+siRNA Mutierte RNA-Matrize in Kombination mit einer sirna gegen die RNA- Matrize 8 Leervektor Kontrolle Die Plasmide wurden uns freundlicherweise von Elisabeth Blackburn zur Verfügung gestellt. Da in allen transduzierten Plasmiden ein GFP-Gen platziert ist, konnten die erfolgreich transduzierten Zellen unter einem Fluoreszenzmikroskop auf Transformationseffizienz bestimmt werden. Die Transduktionseffizienz betrug je nach Plasmid bei den genetisch definierten Plattenepithelkarzinomzellen zwischen 10-60%, bei den genetisch definierten Barrettzellen zwischen 15-50% und bei den genetisch nicht definierten Adenokarzinomzellen zwischen 10-60%. Um die transduzierten Zellen anzureichern, wurden die Zellen 2 Tage nach Infektion an einem FACS-Zellsorter nach GFP sortiert. Um eine Mischpopulation von GFP-positiven und GFP-negativen Zellen möglichst gering zu halten, wurden die Zellen alle zwei bis drei Monate neu sortiert Genetische Inhibition der Telomerase führt zur Reduktion der Telomeraseaktivität und zum Erhalt von Telomeren in genetisch definierten Zellen Im ersten Schritt wurde geprüft, ob die genetische Inhibition der Telomerase mit den Plasmiden 2-7 auch tatsächlich zu einer Reduktion der Telomeraseaktivität führte. Dafür wurde Protein aus den mit den Plasmiden 2-7 transduzierten, genetisch definierten Plattenepithelkarzinomzellen, den genetisch definierten Barrettzellen und den Adenokarzinomzellen sowie den korrelierenden Telomerase-positiven Kontrollzellen gewonnen, und ein TRAP-Assay bei unterschiedlichen Passagen durchgeführt. Unterschiedliche Passagen wurden untersucht, um über eine bestimmte Zeitspanne die Telomeraseinhibition zu beobachten, und deren Stabilität untersuchen zu können. 53


 7 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 8 (Leervektor) Abb. 3.")
 und 204 (3-8) (C), zwischen 222 und 262 (D)) wurden diese mit CHAPS-Lysepuffer lysiert und 10 ng Protein für den TRAP-Assay eingesetzt.")
61 3 Ergebnisse Untersuchungen der Telomeraseaktivität und Telomerlängen in genetisch definierten Plattenepithelkarzinomzellen Wie in Abbildung 3.7 sichtbar, war die Telomeraseaktivität im TRAP-Assay in den genetisch definierten Plattenepithelkarzinomzellen die mit den Kontrollplasmiden 1 und 8 transduziert oder untransduziert waren (mit 0 bezeichnet), wie erwartet sehr stark. Es zeigte sich stabil über viele Passagen (von , Abb. 3.7 A-D), eine leichte Reduktion der Telomeraseaktivität lediglich nach Transduktion mit Plasmid 7. A B C D HI IK OKF6 D1/dnp53/EGFR/myc-Zellen untransduziert 1 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 1 (WT-hTER) 2 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 2 (MT-hTER/AU5) 3 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 3 (MT-hTER/47A) 6 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 8 (Leervektor) Abb. 3.7: Telomeraseaktivität in unterschiedlichen Passagen der Telomerase-inhibierten OKF6 D1/dnp53/EGFR/myc- und Kontrollzellen Nach Ernte der OKF6 D1/dnp53/EGFR/myc-Zellen (Passage 167 (A), 181 (B), 196 (1-2) und 204 (3-8) (C), zwischen 222 und 262 (D)) wurden diese mit CHAPS-Lysepuffer lysiert und 10 ng Protein für den TRAP-Assay eingesetzt. Die radioaktiven PCR-Produkte wurden auf einem Acrylamidgel aufgetrennt, das Gel getrocknet und die radioaktive Strahlung auf einem Röntgenfilm über Nacht bei -80 C detektiert. HI: Hitzeinaktiviert; IK: interne Kontrolle; + positiv Kontrolle aus dem Kit; - negativ Kontrolle (CHAPS-Puffer). Gleichzeitig kam es bei diesen Zellen, die mit dem Plasmid 7 transduziert wurden, zu einer Veränderung der Telomerlänge (Abb. 3.8): Wenige Passagen nach lentiviraler Transduktion bei Passage 167 (Abb. 3.8 A) sind die Telomerlängen, der mit Telomeraseinhibitoren transduzierten, genetisch definierten Plattenepithelkarzinomzellen (OKF6 D1/dnp53/ EGFR/myc), sowie die der Kontrollzellen gleichlang und liegen zwischen 18 und 20 kbp. Die Bande im Southern Blot ist scharf begrenzt, was auf eine Homogenität der Telomerlängen hinweißt. 14 Passagen später, bei Passage 181 (3.8 B) werden die Kontrollzellen, die WThTER (Plasmid 1) überexprimieren sogar länger, als die mit Leervektor transduzierten Kontrollzellen (Plasmid 8). 54
, bleiben in diese Zellen die Telomere zunächst stabil. Was jedoch in den mit Plasmid 7 transduzierten Zellen im Vergleich zu den anderen transduzierten bzw.")
 noch deutlicher. Hier sind die Telomerlängen sehr heterogen und reichen von ca. 8 kbp bis 20 kbp.")
 3 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 3 (MT-hTER/47A) 6 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OKF6 D1/dnp53/EGFR/myc-Zellen")
,")
62 3 Ergebnisse Wenn man die Telomere in den mit Plasmid 7 transduzierten Zellen näher analysiert, bei denen die Telomeraseaktivität reduziert war (Abb. 3.7), bleiben in diese Zellen die Telomere zunächst stabil. Was jedoch in den mit Plasmid 7 transduzierten Zellen im Vergleich zu den anderen transduzierten bzw. Kontrollzellen zusätzlich sichtbar wird ist, dass ein Schmier, d.h. eine heterogene Telomerlänge entsteht. Dies ist charakteristisch für den ALT- Mechanismus. Diese Heterogenität wird in späteren Passagen (3.8 C) noch deutlicher. Hier sind die Telomerlängen sehr heterogen und reichen von ca. 8 kbp bis 20 kbp. A B C D 21 kbp 5 kbp OKF6 D1/dnp53/EGFR/myc-Zellen untransduziert 1 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 1 (WT-hTER) 2 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 2 (MT-hTER/AU5) 3 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 3 (MT-hTER/47A) 6 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OKF6 D1/dnp53/EGFR/myc-Zellen Plasmid 8 (Leervektor) Abb. 3.8: Telomerlängenbestimmung anhand TRF-Analysen in Telomerase-inhibierten OKF6 D1/dnp53/EGFR/myc-Zellen und Kontrollzellen Nach DNA-Extraktion aus den OKF6 D1/dnp53/EGFR/myc-Zellen (Passage 167 (A), 181 (B), 196 (1-2) und 204 (3-8) (C), zwischen 222 und 262 (D)) wurden 5 µg DNA mit den Enzymen HinfI und RsaI verdaut, auf einem 0,5% Agarosegel aufgetrennt, auf eine Hybond XL-Membran transferiert und mit einer radioaktiv-markierten Sonde gegen die Telomere hybridisiert und das Signal auf einem Röntgenfilm detektiert. Weitere 40 Passagen später (3.8 D) wird diese noch deutlicher. Die Telomerlängen reichen von 5 kbp bis 18 kbp. Somit könnten die Telomerase-inhibierten, genetisch definierten Plattenepithelkarzinomzellen auf einen alternativen Telomererhaltungsmechanismus zurückgreifen, der sich in höheren Passagen immer stärker ausprägt und somit die Telomerlängen erhält Untersuchung der Telomeraseaktivität und Telomerlängen in den genetisch definierten Barrettzellen Wie auch in den genetisch definierten Plattenepithelkarzinomzellen, wurde die Telomeraseinhibition über viele Passagen bei den mit Telomeraseinhibitoren transduzierten genetisch 55
 mit den Telomeraseinhibitoren transduziert, um zu garantieren, dass die Zellen nur Veränderungen enthalten, die in Kapitel 3.")

 Abb. 3.")

63 3 Ergebnisse definierten Barrettzellen und Kontrollzellen beobachtet. Diese Zellen wurden bei einer sehr niedrigen Ausgangspassage (Passage 26) mit den Telomeraseinhibitoren transduziert, um zu garantieren, dass die Zellen nur Veränderungen enthalten, die in Kapitel beschrieben wurden, und somit weiterhin als genetisch definiert bezeichnet werden konnten. A B C D HI IK CP-A-Zellen untransduziert 1 CP-A-Zellen Plasmid 1 (WT-hTER) 2 CP-A-Zellen Plasmid 2 (MT-hTER/AU5) 3 CP-A-Zellen Plasmid 3 (MT-hTER/47A) 6 CP-A-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 CP-A-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 CP-A-Zellen Plasmid 8 (Leervektor) Abb. 3.9: Telomeraseaktivität in unterschiedlichen Passagen der Telomerase-inhibierten CP-A- Zellen und Kontrollzellen Nach Ernte der CP-A-Zellen (Passage 32 (A), 50 (B), 86 (C), 109 (D)) wurden diese mit CHAPS- Lysepuffer lysiert und 30 ng an Protein für den TRAP-Assay eingesetzt. Die radioaktiven PCR- Produkte wurden auf einem Acrylamidgel aufgetrennt, das Gel getrocknet und die radioaktive Strahlung auf einem Röntgenfilm über Nacht detektiert. HI: Hitzeinaktiviert; IK: interne Kontrolle. Wie in Abbildung 3.9 sichtbar, kommt es auch hier, ebenso, wie in den zuvor beschrieben genetisch definierten Plattenepithelkarzinomzellen, zu einer Reduktion der Telomeraseaktivität nach Transduktion mit Plasmid 7. Zum Teil kommt es sogar zum fast vollständigen Verschwinden der Telomeraseaktivität in den genetisch definierten Barrettzellen. Diese Inhibition bleibt, wie in Abbildung 3.9 dargestellt, über viele Passagen konstant. Auch hier wurde im nächsten Schritt die Telomerlänge der Kontrollzellen mit denen der Telomerase-inhibierten, genetisch definierten Barrettzellen anhand eines TRFs mit anschließendem Southern Blot verglichen. Drei Passagen nach Transduktion mit den Plasmiden 1-8 (Abbildung 3.10 A) sind die Telomere stabil und liegen bei ca. 12 kbp. 56

.")
 2 CP-A-Zellen Plasmid 2 (MT-hTER/AU5) 3 CP-A-Zellen Plasmid 3")
64 3 Ergebnisse Untersuchungen der Telomerlängen in Passage 50 zeigen nach Transduktion mit Plasmid 1 (WT-hTER) eine Verlängerung der Telomere, die schließlich über 21 kbp liegen. Die Telomerlängen der Telomerase-inhibierten Zellen bleiben zunächst gleichlang wie bei den Kontrollzellen, obwohl im TRAP eine deutliche Reduktion der Telomeraseaktivität nach Transduktion mit Plasmid 7 zu sehen ist. Weitere 36 Passagen später, bei Passage 86 (Abb C) bleiben die Telomere in den Kontrollzellen gleichlang. Dagegen werden die Telomere der CP-A-Zellen, die das Telomerase-inhibitorische Plasmid 7 enthalten, gering kürzer und ein ALT spezifischer Schmier tritt auf (Abb C). Diese Telomerlängenheterogenität bleibt über weitere 23 Passagen konstant (Abb.3.10 D), was wiederum auf einen ALT-Mechanismus hinweist. A B C D 21 bp 5 bp CP-A-Zellen untransduziert 1 CP-A-Zellen Plasmid 1 (WT-hTER) 2 CP-A-Zellen Plasmid 2 (MT-hTER/AU5) 3 CP-A-Zellen Plasmid 3 (MT-hTER/47A) 6 CP-A-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 CP-A-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 CP-A-Zellen Plasmid 8 (Leervektor) Abb. 3.10: Telomerlängenbestimmung anhand TRF-Analysen in Telomerase-inhibierten CP-A- Zellen und Kontrollzellen Nach DNA-Extraktion aus den CP-A-Zellen (Passage 32 (A), 50 (B), 86 (C), 109 (D)) wurden 5 µg DNA mit den Enzymen HinfI und RsaI verdaut, auf einem 0,5% Agarosegel aufgetrennt, dann auf eine Hybond XL-Membran transferiert und mit einer radioaktiv-markierten Sonde gegen die Telomere hybridisiert und das Signal auf einem Röntgenfilm detektiert Untersuchungen der Telomeraseaktivität und Telomerlängen in den genetisch nicht definierten Adenokarzinomzellen Auch in den genetisch nicht definierten Adenokarzinomzellen (OE19) sollten die Telomeraseaktivität und die Telomerlängen nach Transduktion mit den verschiedenen Telomeraseinhibitoren untersucht werden. Hier kam es auch zu einer leichten Reduktion der Telomerase nach lentiviraler Transduktion mit den Telomerase-inhibitorischen Plasmiden 2-7. Am deutlichsten und über viele Passagen hinweg, kam es zu einer Abnahme der Telomeraseaktivität nach Transduktion mit Plasmid 2, wie in Abbildung 3.11 dargestellt. Die Kontrollzellen dagegen zeigten über alle Passagen eine konstante Telomeraseaktivität. 57
 2 OE19-Zellen Plasmid 2 (MT-hTER/AU5) 3 OE19-Zellen Plasmid 3 (MT-hTER/47A) 6 OE19-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OE19-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OE19-Zellen")
, 70 (B), 80 (C), 100 (D)) wurden diese mit")

.")
65 3 Ergebnisse A B C D HI IK OE19-Zellen untransduziert 1 OE19-Zellen Plasmid 1 (WT-hTER) 2 OE19-Zellen Plasmid 2 (MT-hTER/AU5) 3 OE19-Zellen Plasmid 3 (MT-hTER/47A) 6 OE19-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OE19-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OE19-Zellen Plasmid 8 (Leervektor) Abb. 3.11: Telomeraseaktivität in unterschiedlichen Passagen der Telomerase-inhibierten OE19-Zellen und Kontrollzellen Nach Ernte der OE19-Zellen (Passage 53 (A), 70 (B), 80 (C), 100 (D)) wurden diese mit CHAPS- Lysepuffer lysiert und 30 ng an Protein für den TRAP-Assay eingesetzt. Die radioaktiven PCR- Produkte wurden auf einem Acrylamidgel aufgetrennt, das Gel getrocknet und die radioaktive Strahlung auf einem Röntgenfilm über Nacht detektiert. HI: Hitzeinaktiviert; IK: interne Kontrolle. Die Telomerlängen der Adenokarzinomzellen sind von jeher sehr heterogen und liegen zwischen 7 und 12 kbp. Diese Heterogenität bleibt über die jeweils untersuchten Passagen erhalten (Abb. 3.12). In Passage 53, kurz nach Transduktion der Adenokarzinomzellen mit den Telomerase-inhibitorischen Plasmide 2-7 sind die Telomerlängen gleichlang lang wie in den Kontrollzellen (untransduziert, Plasmide 1 und 8). Wie in Abbildung 3.12 dargestellt, nimmt die Telomerlänge der Kontrollzellen, die mit Plasmid 1 transduziert sind und Wildtyp RNA-Matrize enthalten, leicht zu. Diese Verlängerung ist jedoch nicht so stark ausgeprägt, wie bei den genetisch definierten Plattenepithelkarzinomzellen und den genetisch definierten Barrettzellen. Die Telomerlängen der Adenokarzinomzellen wird von Passage nach Transduktion mit Plasmid 2 stetig kürzer (< 5 kbp), wie in Abbildung 3.12 gezeigt. Die schon initial bestehende Heterogenität bleibt erhalten. Dies korreliert gut mit der Abnahme der Telomeraseaktivität im TRAP-Assay. 58

, 70 (B), 80 (C), 100 (D)) wurden 5 µg")


66 3 Ergebnisse A B C D 21 kbp 5 kbp OE19-Zellen untransduziert 1 OE19-Zellen Plasmid 1 (WT-hTER) 2 OE19-Zellen Plasmid 2 (MT-hTER/AU5) 3 OE19-Zellen Plasmid 3 (MT-hTER/47A) 6 OE19-Zellen Plasmid 6 (MT-hTER/AU5+siRNA) 7 OE19-Zellen Plasmid 7 (MT-hTER/47A+siRNA) 8 OE19-Zellen Plasmid 8 (Leervektor) Abb. 3.12: Telomerlängenbestimmung anhand TRF-Analysen in Telomerase-inhibierten OE19- Zellen und Kontrollzellen Nach DNA-Extraktion aus den OE19-Zellen (Passage 53 (A), 70 (B), 80 (C), 100 (D)) wurden 5 µg DNA mit den Enzymen HinfI und RsaI verdaut, auf einem 0,5% Agarosegel aufgetrennt, dann auf eine Hybond XL-Membran transferiert und mit einer radioaktiv-markierten Sonde gegen die Telomere hybridisiert. Das Signal wurde mittel einem Röntgenfilm detektiert Zellmorphologische Charakterisierung der Telomerase-inhibierten Zellen Nach erfolgreicher Reduktion der Telomerase durch die Telomerase-inhibitorischen Plasmide, wurden die drei Zelllinien auf ihre Zellmorphologie untersucht. Wie in Abbildung 3.13 gezeigt, unterscheiden sich die Telomerase-inhibierten, GFP-positiven Zellen morphologisch nicht von den untransduzierten und mit Wildtyp Plasmid 1 transduzierten Zellen. Die Zellen teilten sich normal weiter und wiesen auch nach weiteren Zellteilungen keine zellmorphologischen Veränderungen der Seneszenz oder Apoptose auf. Die GFPpositiven Zellen unterschieden sich nur in ihrer Leuchtintensität. Zum Teil kamen sehr stark leuchtende Zellen vor, aber auch sehr schwach leuchtende Zellen, was darauf hinweißt, dass unterschiedlich viel Plasmid pro Zelle aufgenommen wurde (Abbildung 3.13). 59



, CP-A- (Mitte) und")





67 3 Ergebnisse OKF6 D1/dnp53/EGFR/myc-Zellen; untransduziert OKF6 D1/dnp53/EGFR/myc-Zellen; transduziert mit Plasmid 1 OKF6 D1/p53/EGFR/myc-Zellen; transduziert mit Plasmid 7 CPA-Zellen; untransduziert CPA-Zellen; transduziert mit Plasmid 1 CPA-Zellen; transduziert mit Plasmid 7 OE19-Zellen; untransduziert OE19-Zellen; transduziert mit Plasmid 1 OE19-Zellen; transduziert mit Plasmid 2 Abb. 3.13: Fluoreszenzmikroskopische Beispielbilder der lentiviral-transduzierten Zellen und Kontrollzellen OKF6 D1/dnp53/EGFR/myc- (oben), CP-A- (Mitte) und OE19-Zellen (unten) wurden mit verschiedenen lentiviralen Vektoren infiziert. Unter einem Zeiss Fluoreszenzmikroskop wurden die Zellen betrachtet und mit einer 20x Vergrößerung fotografiert. Auf der linken Seite zum Vergleich untransduzierte Zellen Wachstumsverhalten der Telomerase-inhibierten Zellen Nachdem bei den zellmorphologischen Untersuchungen keine Unterschiede zwischen den erfolgreich Telomerase-inhibierten und den Telomerase-positiven Zellen festzustellen waren, wurde im nächsten Schritt das Wachstumsverhalten der untransduzierten mit dem der Telomerase-inhibierten Zellen verglichen. Um das Wachstum in den mit Telomeraseinhibitoren transduzierten, genetisch definierten Plattenepithelkarzinomzellen (Plasmide 2-7) und den korrespondierenden Kontrollzellen (Plasmide 1 und 8) zu untersuchen, wurde zunächst der Anteil der GFP-positiven Zellen an der Gesamtpopulation in zweitägigen 60
68 3 Ergebnisse Rhythmus ausgezählt und dann graphisch dargestellt. Wie man in Abbildung 3.14 sehen kann, nimmt der Anteil der GFP-positiven Zellen nur nach Transduktion mit Plasmid 2 und 6 leicht ab. Die Transduktion der Zellen mit Plasmid 7 führt dagegen lediglich zu einer langsameren GFP-Abnahme. Die mit Plasmid 1 und 8 transduzierten Kontrollzellen zeigen nur eine relativ geringe Abnahme der GFP-Positivität % Prozent Reihe1 Reihe2 Reihe3 Reihe4 Reihe5 Reihe Tage Reihe1 OKF6 D1/dnp53/EGFR/myc Plasmid 1 Reihe2 OKF6 D1/dnp53/EGFR/myc Plasmid 2 Reihe3 OKF6 D1/dnp53/EGFR/myc Plasmid 3 Reihe4 OKF6 D1/dnp53/EGFR/myc Plasmid 6 Reihe5 OKF6 D1/dnp53/EGFR/myc Plasmid 7 Reihe6 OKF6 D1/dnp53/EGFR/myc Plasmid 8 Abb. 3.14: Graphische Darstellung des Wachstumsverhalten der OKF6 D1/dnp53/EGFR/myc- Zellen Alle 2-3 Tage, beginnend nach der ersten Sortierung, wurden die GFP-positiven, Telomeraseinhibierten Zellen und die GFP-negativen Zellen an einem Fluoreszenzmikroskop gezählt und anschließend miteinander dividiert. Um Wachstumskurven der Kontrollzellen (Plasmid 1, 8 und untransduzierte Zellen) und der mit Plasmid 2-7 transduzierten, genetisch definierten Barrettzellen bzw. Adenokarzinomzellen zu erstellen, wurden je 1x 10 5 Zellen ausplattiert, nach 2-3 Tagen trypsiniert und die Zellzahl bestimmt. Der Wert wurde in eine Formel zum Erhalt der Populationsdopplungen eingesetzt und berechnet. Dies wurde 4-6 Mal wiederholt. In den genetisch definierten Barrettkontrollzellen, sowie in den mit den Plasmiden 2-7 transduzierten Zellen, die die Telomerase-inhibitorischen Plasmide enthalten, liegt eine Populationsdopplung zwischen 0,9-1,1 pro Tag (Abb A). Ähnlich sieht es bei den mit den Plasmiden 2-7 transduzierten Adenokarzinomzellen bzw. bei den Adenokarzinomkontrollzellen aus. Auch hier führt die Transduktion der Telomeraseinhibitoren zu keiner signifikanten Reduktion des Zell- 61
69 3 Ergebnisse wachstums. Diese Zellen wuchsen allgemein, verglichen mit den CP-A-Zellen jedoch insgesamt langsamer. Sie machen im Schnitt 0,6-0,7 Populationsdopplungen pro Tag durch (Abb B). A 1,2 1 PD/Tag 0,8 0,6 0,4 CPA_leer CPA_1 CPA_2 CPA_3 CPA_6 CPA_7 CPA_8 0,2 0 1 B 1,2 1 PD/Tag 0,8 0,6 0,4 OE19_leer OE19_1 OE19_2 OE19_3 OE19_6 OE19_7 OE19_8 0,2 0 1 Abb. 3.15: Graphische Darstellung des Wachstumsverhalten der Telomerase-inhibierten CP-A- Zellen (A) und OE19-Zellen (B) Nach Trypsinierung der Zellen wurden je Zelltyp und Konstrukt 1x 10 5 Zellen gezählt und ausgesät. Drei Tage später wurden die Zellen erneut trypsiniert und gezählt und schließlich die Populationsdopplungen (PD) pro Tag errechnet. 62
70 3 Ergebnisse Telomerlängenbestimmung auf Einzelzellbasis mittels Q-FISH-Analyse In TRF-Analysen mit folgendem Southern Blot misst man die Telomerlängen der gesamten DNA einer transduzierten Subpopulation und das Ergebnis ist stark vom Anteil GFP-positiver Zellen abhängig. Deshalb wurde als nächstes eine quantitative Fluoreszenz in situ Hybridisierung durchgeführt. Hier kann auf Einzelzellebene die Telomerlänge der prämalignen und malignen Zellen untersucht werden. Dabei wurden, nach erfolgreicher Metaphasenpräparation, die Telomere mittels einer Fluoreszenz-markierten Sonde, komplementär zu den Telomeren, sichtbar gemacht. Anhand ihrer Leuchtintensität konnten so die Telomerlängen bestimmt werden, wobei die Telomerlänge mit der Fluoreszenzstärke korreliert. Da in ALT-positiven Zellen auch Telomer-freie Enden neben sehr langen Telomeren vorkommen können, die in TRF-Analysen so nicht erfasst werden können, ist die Q-FISH-Analyse hier besonders gut geeignet. Zunächst wurde der Q-FISH an den immortalisierten ösophagealen Keratinozyten (EPChTERT-Zellen) etabliert. An den Beispielbildern in Abbildung 3.16 A und B ist sehr deutlich zu sehen, dass in den Kontrollzellen die Telomere ein sehr homogenes Bild aufweisen. Das Signal an den Telomeren (rot) ist an allen Chromatiden nahezu gleich stark. Auffällig ist dagegen, dass in den Telomerase-inhibierten immortalisierten ösophagealen Keratinozyten, transduziert mit Plasmid 6, die Telomere sehr heterogen sind (Abbildung 3.16 C und D). Es sind an den 4 Telomerenden eines jeweiligen Metaphasenchromosoms sehr starke Telomersignale sichtbar, aber auch extrem schwache bis überhaupt keine Signale mehr nachweisbar. In der Zusammenfassung der Q-FISH-Analyse, in Abbildung 3.17, wird deutlich, dass in den Telomerase-inhibierten, immortalisierten, ösophagealen Keratinozyten (Plasmid 3 und 6), verglichen mit den Kontrollzellen (untransduziert, Plasmide 1 und 8) fast ausschließlich heterogene Telomerlängen nachzuweisen waren. Der Vergleich mit den Kontrollzellen war hier wichtig, da zur Markierung der Telomere an Metaphasen keine intakten Zellen mehr vorhanden sind und somit in diesem Stadium die GFP-positiven nicht mehr von den GFPnegativen Zellen unterschieden werden konnten. 63


 und die Telomere mit einer Cy3-markierten Sonde (rot), die komplementär zu der")
71 3 Ergebnisse A B Abb A und B: Beispielbilder der Telomerlängenbestimmung in EPC-hTERT-Zellen mit Hilfe einer Q-FISH-Analyse EPC-hTERT Zellen wurden ausgesät und am nächsten Tag 6 h mit Colcemid behandelt, um die Zellen in der Metaphase zu arretieren. Nach einer KCl-Behandlung und anschließender Fixierung hatten die Zellen Zeit anzuschwellen, bevor sie auf einen Objektträger getropft und somit zum Platzen gebracht wurden. Die so erhaltenen Metaphasen wurden mit DAPI angefärbt (blau) und die Telomere mit einer Cy3-markierten Sonde (rot), die komplementär zu der Telomersequenz war, hybridisiert. (A und B) EPC-hTERT Plasmid 1 und untransduzierte Kontrollzellen. (C und D) EPChTERT transduziert mit Plasmid 6 und Plasmid 3 (nächste Seite). Die Bilder wurden an einem Fluoreszenzmikroskop mit einer 63 fachen Vergrößerung aufgenommen. Telomer-freie Enden in C und D werden durch einen weißen Pfeil markiert. 64






72 3 Ergebnisse C D Abb C und D: Beispielbilder der Telomerlängenbestimmung in EPC-hTERT-Zellen mit Hilfe einer Q-FISH-Analyse Legende Seite 64 65
73 3 Ergebnisse Prozent heterogene Telomere homogene Telomere nicht auswertbar Plasmide Abb. 3.17: Zusammenfassung der Q-FISH-Analyse mittels Histogramm der mit Telomeraseinhibitoren transduzierten EPC-hTERT-Zellen und Kontrollzellen. (0) EPC-hTERT untransduziert, (1) EPC-hTERT Plasmid 1 (WT-hTER), (3) EPC-hTERT Plasmid 3 (MT-hTER/47A), (6) EPC-hTERT Plasmid 6 (MT-hTER/AU5+siRNA), (8) EPC-hTERT Plasmid 8 (Leervektor). Zunächst wurden alle Metaphasen betrachtet und nur diejenigen ausgewertet, deren Chromosomen gut verteilt waren. Im nächsten Schritt wurden das Telomersignal in drei Kategorien eingeteilt: heterogenes Signal ( heterogene Telomere), homogenes Signal ( homogene Telomere) und Signale, die nicht auswertbar waren. Nachdem die Q-FISH-Methode an den EPC-hTERT-Zellen etabliert war, wurde sie an den genetisch definierten Plattenepithelkarzinomzellen, den genetisch definierten Barrettzellen und an den genetisch nicht definierten Adenokarzinomzellen durchgeführt, um auf Einzelzellebene die Telomerlängen in diesen Zellen zu untersuchen. Auch bei den genetisch definierten Plattenepithelkarzinomzellen (OKF6 D1/dnp53/EGFR/ myc), den genetisch definierten Barrettzellen (CP-A) und den genetisch undefinierten Adenokarzinomzellen (OE19) sind die Telomerlängen zunächst homogen und das Signal bei allen Kontrollzellen sehr stark (Abbildungen 3.18, 3.20 und 3.22 A und B). Anders sieht es bei den mit Telomeraseinhibitoren behandelten Zellen aus. In den genetisch definierten Plattenepithelkarzinomzellen, die im TRAP-Assay eine reduzierte Telomeraseaktivität nach Transduktion mit Plasmid 7 zeigten, findet man in der Q-FISH-Analyse sehr heterogene Telomere, mit sehr starken Signalen und gleichzeitig sehr schwachen bzw. keinen Signalen innerhalb der Telomerenden der Chromosomen. Dies korreliert mit den Telomerlängen aus den TRF-Analysen, bei denen an der Gesamtpopulation und gesamt DNA ebenfalls heterogenen Telomerlängen aufgezeigt werden konnten. 66



 und die Telomere mit")

74 3 Ergebnisse A B Abb A und B: Beispielbilder der Telomerlängenbestimmung in OKF6 D1/p53/EGFR/myc- Zellen mithilfe einer Q-FISH-Analyse Die mit Telomerase-inhibierten OKF6 D1/dnp53/EGFR-Zellen und Kontrollzellen wurden ausgesät und am nächsten Tag 6 h mit Colcemid behandelt um die Zellen in der Metaphase zu arretieren. Nach der KCl-Behandlung und anschließender Fixierung wurden die angeschwollenen Zellen auf einen Objektträger getropft und somit zum Platzen gebracht. Die so erhaltenen Metaphasen wurden mit DAPI angefärbt (blau) und die Telomere mit einer Cy3-markierten Sonde (rot), die komplementär zu der Telomersequenz war, hybridisiert. (A und B) OKF6 D1/dnp53/EGFR/myc Plasmid 8 und untransduzierte Kontrollzellen. (C und D) OKF6 D1/dnp53/EGFR/myc transduziert mit Plasmid 7. Ein weißer Pfeil demonstriert Telomer-freie Enden in C und D. 67




75 3 Ergebnisse C D Abb C und D: Beispielbilder der Telomerlängenbestimmung in OKF6 D1/p53/EGFR/myc- Zellen mithilfe einer Q-FISH-Analyse Legende Seite 67 68
76 3 Ergebnisse Prozent heterogene Telomere homogene Telomere nicht auswertbar Plasmide Abb. 3.19: Zusammenfassung der Q-FISH-Analyse im Histogramm der mit Telomeraseinhibitoren transduzierten OKF6 D1/dnp53/EGFR/myc-Zellen und Kontrollzellen. (0) OKF6 D1/dnp53/EGFR/myc untransduziert, (1) OKF6 D1/dnp53/EGFR/myc Plasmid 1, (2) OKF6 D1/dnp53/EGFR/myc Plasmid 2, (3) OKF6 D1/dnp53/EGFR/myc Plasmid 3, (6) OKF6 D1/dnp53/EGFR/myc Plasmid 6, (7) OKF6 D1/dnp53/EGFR/myc Plasmid 7, (8) OKF6 D1/dnp53/EGFR/myc Plasmid 8. Zunächst wurden alle Metaphasen betrachtet und nur diejenigen ausgewertet, deren Chromosomen gut verteilt waren. Im nächsten Schritt wurden das Telomersignal in drei Kategorien eingeteilt: heterogenes Signal ( heterogene Telomere), homogenes Signal ( homogene Telomere) und Signale, die nicht auswertbar waren. In Abbildung 3.19 sind die Ergebnisse der Q-FISH-Analyse der definierten Plattenepithelkarzinomzellen im Diagramm zusammengefasst. In den Kontrollzellen (Plasmide 1 und 8 und untransduziert) lassen sich hier auch hin und wieder heterogene Telomerlängen finden, genau wie in den Telomerase-inhibierten Zellen auch Zellen mit homogenen Telomerlängen vorkommen können. Ebenso konnten in den genetisch definierten Barrettzellen heterogene Telomerlängen in den Telomerase-inhibierten Zellen nachgewiesen werden (Abbildung 3.20 C und D). In den Kontrollzellen dagegen blieben die Telomerlängen homogen (Abbildung 3.20 A und B). 69



 und die Telomere mit einer")
77 3 Ergebnisse A B Abb A und B: Beispielbilder der Telomerlängenbestimmung in CP-A-Zellen mittels einer Q-FISH-Analyse Die mit Telomerase-inhibierten CP-A-Zellen und Kontrollzellen wurden ausgesät und am nächsten Tag 6 h mit Colcemid behandelt um die Zellen in der Metaphase zu arretieren. Nach der KCl- Behandlung und anschließender Fixierung wurden die angeschwollenen Zellen auf einen Objektträger getropft und somit zum Platzen gebracht. Die so erhaltenen Metaphasen wurden mit DAPI angefärbt (blau) und die Telomere mit einer Cy3-markierten Sonde (rot), die komplementär zu der Telomersequenz war, hybridisiert. (A und B) mit Leervektor transduzierte CP-A Kontrollzellen. (C und D) CP-A transduziert mit Plasmid 7. 70


78 3 Ergebnisse C D Abb C und D: Beispielbilder der Telomerlängenbestimmung in CP-A-Zellen mittels einer Q-FISH-Analyse Legende Seite 70 71
79 3 Ergebnisse In der Zusammenfassung der Daten im Histogramm wird deutlich (Abb. 3.21), dass in den Zellen, die mit Plasmid 7 transduziert wurden, die meisten, also 70% der ausgewerteten Metaphasen heterogene Telomere aufzeigen. Dies korreliert mit den Ergebnissen aus dem TRAP-Assay, bei dem es zu einer Abnahme der Telomeraseaktivität in CP-A-Zellen, die mit Plasmid 7 transduziert waren, kam, sowie den Daten der TRF-Analysen, die eine zunehmende Heterogenität der Telomerlängen über die Gesamtpopulation/DNA zeigten Prozent heterogene Telomere homogene Telomere nicht auswertbar Plasmide Abb. 3.21: Zusammenfassung der Q-FISH-Analyse im Histogramm der mit Telomeraseinhibitoren transduzierten CP-A-Zellen und Kontrollzellen. (0) CP-A untransduziert, (1) CP-A Plasmid 1, (2) CP-A Plasmid 2, (3) CP-A Plasmid 3, (6) CP-A Plasmid 6, (7) CP-A Plasmid 7, (8) CP-A Plasmid 8. Zunächst wurden alle Metaphasen betrachtet und nur diejenigen ausgewertet, deren Chromosomen gut verteilt waren. Im nächsten Schritt wurden das Telomersignal in drei Kategorien eingeteilt: heterogenes Signal ( heterogene Telomere), homogenes Signal ( homogene Telomere) und Signale, die nicht auswertbar waren. Eine Schwierigkeit der Q-FISH-Analyse der Telomerase-inhibierten OE19-Zellen (Plasmid 2 und 3) war, dass die Telomere schon vor einer Transduktion in der TRF-Analyse sehr kurz waren, und deshalb schwierig ein Signal in der Q-FISH-Hybridisierung überhaupt nachzuweisen war. Somit musste die Belichtungszeit ungewöhnlich hoch gesetzt werden. Nichtsdestotrotz kann man sagen, dass die Telomerlängen in den Zellen, in denen im TRAP-Assay die Telomerase reduziert war (Plasmid 2 und 3) zwar viel kürzer als in den Kontrollzellen sind, ihre Heterogenität aber konstant beibehalten (Abb.3.22 C und D und Abb. 3.23). 72



, die komplementär zu der Telomersequenz war, hybridisiert.")
80 3 Ergebnisse A B Abb A und B: Beispielbilder der Telomerlängenbestimmung in OE19-Zellen mittels einer Q-FISH-Analyse Die mit Telomerase-inhibierten OE19-Zellen und Kontrollzellen wurden ausgesät und am nächsten Tag 6 h mit Colcemid behandelt um die Zellen in der Metaphase zu arretieren. Nach der KCl- Behandlung und anschließender Fixierung wurden die angeschwollenen Zellen gut resuspendiert und auf einen Objektträger getropft und somit zum Platzen gebracht. Die so erhaltenen Metaphasen wurden mit DAPI angefärbt (blau) und die Telomere mit einer Cy3-markierten Sonde (rot), die komplementär zu der Telomersequenz war, hybridisiert. (A und B) OE19-Zellen transduziert mit Plasmid 8 und untransduzierte OE19 Kontrollzellen. (C und D) OE19 transduziert mit Plasmid 2 und Plasmid 3. Ein weißer Pfeil demonstriert Telomer-freie Enden in C und D. 73

81 3 Ergebnisse C D Abb C und D: Beispielbilder der Telomerlängenbestimmung in OE19-Zellen mittels einer Q-FISH-Analyse Legende Seite 73 74
82 3 Ergebnisse Prozent heterogene Telomere homogene Telomere nicht auswertbar Plasmide Abb. 3.23: Zusammenfassung der Q-FISH-Analyse im Histogramm der mit Telomeraseinhibitoren transduzierten OE19-Zellen und Kontrollzellen. (0) OE19 untransduziert, (1) OE19 Plasmid 1, (2) OE19 Plasmid 2, (3) OE19 Plasmid 3, (6) OE19 Plasmid 6, (7) OE19 Plasmid 7, (8) OE19 Plasmid 8. Zunächst wurden alle Metaphasen betrachtet und nur diejenigen ausgewertet, deren Chromosomen gut verteilt waren. Im nächsten Schritt wurden das Telomersignal in drei Kategorien eingeteilt: heterogenes Signal ( heterogene Telomere), homogenes Signal ( homogene Telomere) und Signale, die nicht auswertbar waren. Die Werte wurden in einer Excel-Tabelle aufgelistet und ein Diagramm erstellt Nachweis von Rekombinationsereignissen an Telomeren zwischen Tochterchromatiden anhand von Chromosomen-Orientierungs-Fluoreszenz in situ Hybridisierung. Da in den vorherigen Abschnitten gezeigt werden konnte, dass die genetisch definierten Zellen, trotz verminderter Telomeraseaktivität, ihre Telomere über ALT erhalten können, sollte im nächsten Schritt überprüft werden, ob es in diesen zu einem Austausch von Schwesterchromatiden kommt. Um das zu untersuchen, wurde der so genannte Chromosomen-Orientierungs-FISH durchgeführt (CO-FISH). Dabei wird der neu replizierte DNA-Strang von Metaphasenchromosomen abgebaut, was zu einer Einzelstrang-Ziel-DNA mit entweder einem C-reichen oder G-reichen Telomerstrang führt. Im nächsten Schritt wird eine Hybridisierung mit spezifischen Sonden, entweder gegen den C- (grün) bzw. G-reichen (rot) Strang durchgeführt. Kommt es zu einer Rekombination zwischen Chromatiden bzw. Schwesterchromatiden, findet man an den Telomeren eine Kolokalisation von beiden Sonden, was zu einer Gelbfärbung führt. Wie man jedoch bei den mit Plasmid 7 transduzierten genetisch definierten Plattenepithelkarzinomzellen und genetisch definierten Barrettzellen sehen kann (Abb B und D) kommt es trotz nachgewiesener heterogenen Telomerlängen im TRF und Q-FISH zu keiner erhöhten Rekombination zwischen Schwesterchromatiden, verglichen mit den Kontrollzellen 75






")


83 3 Ergebnisse (Abb A und C). Somit liegt hier ein ALT-Mechanismus vor, der nicht direkt auf einem Austausch von Chromatiden bzw. Schwesterchromatiden beruht. A B Abb A und B: Chromosomen Orientierungs-FISH an OKF6 D1/dnp53/EGFR/myc-Zellen Nach der Metaphasenpräparation folgte eine Hybridisierung der Telomere mit einer strangspezifischen Sonde entweder gegen den C-reichen (rot) der den G-reichen (grün) Strang mit einer Cy3- bzw. FITC-markierten PNA-Sonde. Die Chromosomen wurden mit DAPI (blau) angefärbt. (A) OKF6 D1/dnp53/EGFR/myc untransduziert (B) OKF6 D1/dnp53/EGFR/myc Plasmid 7 (C) CP-A untransduziert (D) CP-A Plasmid 7. 76


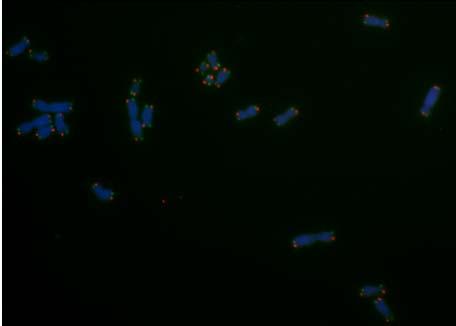
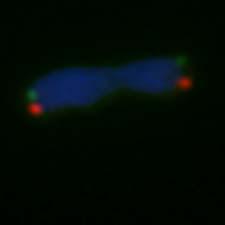

84 3 Ergebnisse C D Abb C und D: Chromosomen Orientierungs-FISH an CP-A-Zellen Legende Seite 76 77
85 3 Ergebnisse ALT-assoziierte PML-Bodies (APBs) in genetisch definierten immortalisierten und maligne transformierten Zellen sowie in nicht definierten Tumorzellen Eine weitere Methode, die indirekt einen ALT-Mechanismus nachweisen kann, ist die so genannte ALT-assoziierte PML-Body-Analyse. Hierbei kommt eine indirekte Immunfluoreszenz zum Einsatz. Von APBs spricht man, wenn PML-Bodies mit den Telomerbindeproteinen TRF1 und TRF2, Telomer-DNA und u.u. mit Proteinen, die bei der homologen Rekombination eine Rolle spielen, wie z.b. Rad50, in einer Zelle kolokalisieren. In dieser Arbeit wurden die untransduzierten Kontrollzellen sowie die mit Telomeraseinhibitoren transduzierten, genetisch definierten Plattenepithelkarzinomzellen, die genetisch definierten Barrettzellen sowie die Adenokarzinomzellen auf Kolokalisationen von TRF2 und PML-Bodies untersucht. Bei Anwendung dieser Methode kamen bei den transduzierten Zellen drei verschiedene Fluoreszenzfarbstoffe zum Einsatz. Die erfolgreich transduzierten Zellen waren neben den beiden anzufärbenden Proteinen TRF2 und PML zusätzlich GFPpositiv. Daher war eine digitalen Umfärbung der Fluoreszenzen notwendig. Somit wurde GFP als blaue, TRF2 als grüne und PML als rote Fluoreszenz dargestellt. Während bei den untransduzierten Zellen, die GFP-negativ waren, die Kolokalisation (gelb) gut sichtbar war, war die Auswertung der transduzierten, GFP-positiven Zellen durch die zusätzliche blaue Fluoreszenz schwieriger. Ein weiteres Problem war, dass blaue Fluoreszenz mit grüner überlappt, und somit der Hintergrund der grünen Fluoreszenz verstärkt wird, was zu einem insgesamt verstärkten Hintergrund führte. Wie an den Beispielbildern in Abbildung 3.25 und in dem Histogramm in Abbildung 3.27 dargestellt, traten APBs in den Telomerase-inhibierten genetisch definierten Plattenepithelkarzinomzellen (Plasmid 7) in einer höheren Frequenz auf, als in den untransduzierten Kontrollzellen (Abb B) und den mit dem Leervektor transduzierten Zellen (Plasmid 8). Wie im Diagramm dargestellt, haben die untransduzierten- und die mit Plasmid 8 transduzierten Kontrollzellen 79% bzw. 84% an Zellen, die APB negativ sind. Dagegen kamen, in den mit Plasmid 7 transduzierten Zellen, bei denen die Telomeraseaktivität reduziert ist, nur 65% APB-negative Zellen vor, dagegen traten in 30% der Zellen mindestens ein APB pro Zelle und sogar in mehr als 7% der Zellen zwei oder mehr APBs pro Zelle auf. Auffällig war, dass in den Kontrollzellen, die mit Plasmid 1 (WT-hTER-Variante) transduziert waren, ebenso wie in den Telomerase-inhibierten Zellen, vermehrt APBs pro Zelle vorkamen. 78
 OKF6")




86 3 Ergebnisse A B C Abb. 3.25: ALT-assoziierte PML-Bodies in OKF6 D1/dnp53/EGFR/myc-Zellen (A) OKF6 D1/dnp53/EGFR/myc-Kontrollzellen (Plasmid 1) und (B) untransduzierte Zellen bzw. (C) Telomerase-inhibierte OKF6 D1/dnp53/EGFR/myc-Zellen (Plasmid 7) wurden auf Deckgläsern ausgesät, am nächsten Tag fixiert, permeabilisiert, unspezifische Bindungen blockiert und mit einen anti-trf2-antikörper über Nacht inkubiert. Am nächsten Tag wurde der anti-pml-antikörper dazugegeben und 4 h inkubiert. Als 2. Antikörper wurden ein anti-maus Alexa594 und ein anti-ziege Cy3-Antikörper verwendet. Die Proben wurden an einem konfokalen Mikroskop analysiert und fotografiert (40x Vergrößerung). Blau: GFP; rot: PML-Bodies; grün: TRF2 79

 Telomeraseinhibierte CP-A-Zellen (Plasmid 7) wurden auf Deckgläsern ausgesät, am")


87 3 Ergebnisse A B C Abb. 3.26: Indirekte Immunfluoreszenz zum Nachweis von ALT-assoziierte PML-Bodies in CP- A-Zellen (A) CP-A-Kontrollzellen (Plasmid 1) und (B) untransduzierte CP-A-Zellen bzw. (C) Telomeraseinhibierte CP-A-Zellen (Plasmid 7) wurden auf Deckgläsern ausgesät, am nächsten Tag fixiert, permeabilisiert, unspezifische Bindungen blockiert und mit einen anti-trf2-antikörper über Nacht inkubiert. Am nächsten Tag wurde der anti-pml-antikörper dazugegeben und 1 h inkubiert. Als 2. Antikörper wurden ein anti-maus Alexa594 und ein anti-ziege Cy3-Antikörper verwendet. Die Proben wurden an einem konfokalen Mikroskop analysiert und fotografiert (40x Vergrößerung). Blau: GFP; rot: PML-Bodies; grün: TRF2 80
88 3 Ergebnisse A Prozent O APBs 1 APB 2 APBs 3 APBs 5 APBs Plasmide B Prozent APBs 1 APB 2 APBs 3 APBs 4 APBs Plasmide Abb. 3.27: Histogramm der indirekten Immunfluoreszenz (APBs) in genetisch definierten Zellen. (A) Telomerase-inhibierte OKF6 D1/dnp53/EGFR/myc- und Kontrollzellen, (B) Telomerase-inhibierte CP-A-Zellen und Kontrollzellen. Die Auswertung erfolgte mit dem LSM Image Browser. Dabei wurden in den GFP-positiven Zellen und den untransduzierten Kontrollzellen APBs gezählt und die Werte (0 APBs, 1 APB, 2 APBs, 3 APBs, 4 APBs) in einer Excel-Tabelle dokumentiert und graphisch dargestellt. Ebenso, wie in den genetisch definierten ösophagealen Plattenepithelkarzinomzellen, kam es in den genetisch definierten Telomerase-inhibierten Barrettzellen zu einem erhöhten Auftreten an APBs verglichen mit den untransduzierten- und mit Plasmid 8 transduzierten 81


89 3 Ergebnisse Kontrollzellen (Abb und 3.27 B). In den mit Plasmid 7 transduzierten genetisch definierten Barrettzellen, bei denen die Telomeraseaktivität runterreguliert war, sind nur 55% der Zellen APB-frei, jedoch in 45% traten ein oder mehrere APBs pro Zelle auf. Auch hier kamen in 47% der mit Plasmid 1 transduzierten Kontrollzellen ein oder mehr APBs pro Zelle vor (Abb B). Insgesamt kamen in Durchschnitt mehr APBs in den Barrett-Zellen vor als in den Plattenepithelkarzinomzellen. In den Adenokarzinomzellen traten sowohl in den Telomerase-inhibierten Zellen sowie den Kontrollzellen nicht signifikant häufig APBs auf (Abb und 3.29). A B Abb A und B: Indirekte Immunfluoreszenz zum Nachweis von ALT-assoziierte PML- Bodies in OE19-Zellen OE19-Kontrollzellen (Plasmid 1) (A) und untransduzierte OE19-Zellen (B) bzw. Telomerase-inhibierte CP-A-Zellen (Plasmid 7) (C, nächste Seite) wurden auf Deckgläsern ausgesät, am nächsten Tag fixiert, permeabilisiert, unspezifische Bindungen blockiert und mit einen anti-trf2-antikörper über Nacht inkubiert. Am nächsten Tag wurde der anti-pml-antikörper dazugegeben und 1h inkubiert. Als 2. Antikörper wurden ein anti-maus Alexa594- und ein anti-ziege Cy3-Antikörper verwendet. Die Proben wurden an einem konfokalen Mikroskop analysiert und fotografiert (40x Vergrößerung). Blau: GFP; rot: PML-Bodies; grün: TRF2 82


90 3 Ergebnisse C Abb C: Indirekte Immunfluoreszenz zum Nachweis von ALT-assoziierte PML-Bodies in OE19-Zellen Legende Seite Prozent APBs 1 APB 2 APBs Plasmide Abb. 3.29: Histogramm der indirekten Immunfluoreszenz (APBs) in genetisch nicht definierten Zellen Die Auswertung erfolgte mit dem LSM Image Browser Programm. Dabei wurden in den GFP-positiven Telomerase-inhibierten Zellen und den untransduzierten Kontrollzellen APBs gezählt und die Werte (0 APBs, 1 APB, 2 APBs, 3 APBs, 4 APBs) in einer Excel-Tabelle dokumentiert und graphisch dargestellt. 83
KV: DNA-Replikation Michael Altmann
 Institut für Biochemie und Molekulare Medizin KV: DNA-Replikation Michael Altmann Herbstsemester 2008/2009 Übersicht VL DNA-Replikation 1.) Das Zentraldogma der Molekularbiologie 1.) Semikonservative Replikation
Institut für Biochemie und Molekulare Medizin KV: DNA-Replikation Michael Altmann Herbstsemester 2008/2009 Übersicht VL DNA-Replikation 1.) Das Zentraldogma der Molekularbiologie 1.) Semikonservative Replikation
Entstehung von menschlichen Tumorzellen aufgrund bestimmter genetischer Elemente
 Entstehung von menschlichen Tumorzellen aufgrund bestimmter genetischer Elemente Einleitung und Begriffserklärung Krebsentstehung: Zellen ändern normales Entwicklungsprogramm und vermehren sich unkontrolliert
Entstehung von menschlichen Tumorzellen aufgrund bestimmter genetischer Elemente Einleitung und Begriffserklärung Krebsentstehung: Zellen ändern normales Entwicklungsprogramm und vermehren sich unkontrolliert
Marianti Manggau. Untersuchungen zu Signalwegen von SPP in humanen Keratinozyten
 Marianti Manggau Untersuchungen zu Signalwegen von SPP in humanen Keratinozyten Die Deutsche Bibliothek - CIP-Einheitsaufnahme Manggau, Marianti: Untersuchungen zu Signalwegen von SPP in humanen Keratinozyten
Marianti Manggau Untersuchungen zu Signalwegen von SPP in humanen Keratinozyten Die Deutsche Bibliothek - CIP-Einheitsaufnahme Manggau, Marianti: Untersuchungen zu Signalwegen von SPP in humanen Keratinozyten
6. DNA -Bakteriengenetik
 6. DNA -Bakteriengenetik Konzepte: Francis Crick DNA Struktur DNA Replikation Gentransfer in Bakterien Bakteriophagen 2. Welcher der folgenden Sätze entspricht der Chargaff-Regel? A) Die Menge von Purinen
6. DNA -Bakteriengenetik Konzepte: Francis Crick DNA Struktur DNA Replikation Gentransfer in Bakterien Bakteriophagen 2. Welcher der folgenden Sätze entspricht der Chargaff-Regel? A) Die Menge von Purinen
Krebszelle und Metastasierung. Sind Krebszellen. unsterblich? Basiskurs Krebswissen 2016
 Sind Krebszellen Basiskurs Krebswissen 2016 unsterblich? 1 Berger, Walter Univ. Berger, Klinik Institut Innere für Medizin Krebsforschung, I, Institut Medizinische für Krebsforschung Universität Wien Aufbau
Sind Krebszellen Basiskurs Krebswissen 2016 unsterblich? 1 Berger, Walter Univ. Berger, Klinik Institut Innere für Medizin Krebsforschung, I, Institut Medizinische für Krebsforschung Universität Wien Aufbau
Stand von letzter Woche
 RUB ECR1 AXR1 Stand von letzter Woche E2 U? E1-like AXR1 Repressor ARF1 Proteasom AuxRE Repressor wird sehr schnell abgebaut notwendig für Auxinantwort evtl. Substrat für SCF Identifikation des SCF-Ubiquitin
RUB ECR1 AXR1 Stand von letzter Woche E2 U? E1-like AXR1 Repressor ARF1 Proteasom AuxRE Repressor wird sehr schnell abgebaut notwendig für Auxinantwort evtl. Substrat für SCF Identifikation des SCF-Ubiquitin
Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR)
") PCR Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR) von Kary B. Mullis entwickelt (1985) eigentlich im Mai 1983 bei einer nächtlichen Autofahrt erstes erfolgreiches Experiment am 16.12.1983
PCR Genisolierung in 2 Stunden : Die Polymerase-Ketten-Reaktion (PCR) von Kary B. Mullis entwickelt (1985) eigentlich im Mai 1983 bei einer nächtlichen Autofahrt erstes erfolgreiches Experiment am 16.12.1983
Klonierung von Säugern durch Zellkerntransfer
 Klonierung von Säugern durch Zellkerntransfer Gentechnik und Genomics WiSe 2007/2008 Kristian M. Müller Institut für Biologie III Albert-Ludwigs-Universität Freiburg Können differenzierte Zellen einen
Klonierung von Säugern durch Zellkerntransfer Gentechnik und Genomics WiSe 2007/2008 Kristian M. Müller Institut für Biologie III Albert-Ludwigs-Universität Freiburg Können differenzierte Zellen einen
2. Übung: Chromosomentheorie
 Konzepte: 2. Übung: Chromosomentheorie Mitose/Meiose Geschlechtschromosomale Vererbung Chromosomentheorie Regeln zur Vererbung Autosomal rezessiv: - Merkmal tritt auf in Nachkommen nicht betroffener Eltern
Konzepte: 2. Übung: Chromosomentheorie Mitose/Meiose Geschlechtschromosomale Vererbung Chromosomentheorie Regeln zur Vererbung Autosomal rezessiv: - Merkmal tritt auf in Nachkommen nicht betroffener Eltern
Zelluläre Reproduktion: Zellzyklus. Regulation des Zellzyklus - Proliferation
 Zelluläre Reproduktion: Zellzyklus Regulation des Zellzyklus - Proliferation Alle Zellen entstehen durch Zellteilung Der Zellzyklus kann in vier Haupt-Phasen eingeteilt werden Interphase Zellwachstum;
Zelluläre Reproduktion: Zellzyklus Regulation des Zellzyklus - Proliferation Alle Zellen entstehen durch Zellteilung Der Zellzyklus kann in vier Haupt-Phasen eingeteilt werden Interphase Zellwachstum;
DNA Replikation ist semikonservativ. Abb. aus Stryer (5th Ed.)
") DNA Replikation ist semikonservativ Entwindung der DNA-Doppelhelix durch eine Helikase Replikationsgabel Eltern-DNA Beide DNA-Stränge werden in 5 3 Richtung synthetisiert DNA-Polymerasen katalysieren die
DNA Replikation ist semikonservativ Entwindung der DNA-Doppelhelix durch eine Helikase Replikationsgabel Eltern-DNA Beide DNA-Stränge werden in 5 3 Richtung synthetisiert DNA-Polymerasen katalysieren die
Verbleibende Lebenserwartung von Neugeborenen und 70-Jährigen in Deutschland
 abweichen können (interindividuelle Variabilität). Auch laufen einzelne biologische Alterungsprozesse nicht alle zur gleichen Zeit und auch nicht gleich intensiv ab (intraindividuelle Variabilität). Altern
abweichen können (interindividuelle Variabilität). Auch laufen einzelne biologische Alterungsprozesse nicht alle zur gleichen Zeit und auch nicht gleich intensiv ab (intraindividuelle Variabilität). Altern
Unterrichtsmaterialien in digitaler und in gedruckter Form. Auszug aus:
 Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Die klassische Genetik: T.H. Morgan und seine Experimente mit Drosophila melanogaster Das komplette Material finden Sie hier: Download
Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: Die klassische Genetik: T.H. Morgan und seine Experimente mit Drosophila melanogaster Das komplette Material finden Sie hier: Download
Übung 8. 1. Zellkommunikation. Vorlesung Bio-Engineering Sommersemester 2008. Kapitel 4. 4
 Bitte schreiben Sie Ihre Antworten direkt auf das Übungsblatt. Falls Sie mehr Platz brauchen verweisen Sie auf Zusatzblätter. Vergessen Sie Ihren Namen nicht! Abgabe der Übung bis spätestens 05. 05. 08
Bitte schreiben Sie Ihre Antworten direkt auf das Übungsblatt. Falls Sie mehr Platz brauchen verweisen Sie auf Zusatzblätter. Vergessen Sie Ihren Namen nicht! Abgabe der Übung bis spätestens 05. 05. 08
Etablierung, Validierung und praktische Anwendung einer multiplex real-time RT-PCR zum Nachweis des Rabbit Haemorrhagic Disease Virus
 Aus dem Institut für Virologie der Tierärztlichen Hochschule Hannover und dem Institut für Virusdiagnostik des Friedrich-Loeffler-Instituts, Insel Riems Etablierung, Validierung und praktische Anwendung
Aus dem Institut für Virologie der Tierärztlichen Hochschule Hannover und dem Institut für Virusdiagnostik des Friedrich-Loeffler-Instituts, Insel Riems Etablierung, Validierung und praktische Anwendung
Der EGFR-Signalweg als ein Schlüsselmechanismus in der in vitro Transformation und Telomerasereaktivierung
 I Aus der Abteilung Innere Medizin II Gastroenterologie, Hepatologie, Infektiologie und Endokrinologie der Albert-Ludwigs-Universität, Freiburg im Breisgau Ärztlicher Direktor: Prof. Dr. Drs. h.c. H. E.
I Aus der Abteilung Innere Medizin II Gastroenterologie, Hepatologie, Infektiologie und Endokrinologie der Albert-Ludwigs-Universität, Freiburg im Breisgau Ärztlicher Direktor: Prof. Dr. Drs. h.c. H. E.
SCRIPTUM HIGH PRECISE
 Nur für Forschungszwecke Datenblatt Artikel BS.50.020 = 20 Reaktionen x 50 µl Artikel BS.50.100 = 100 Reaktionen x 50 µl Artikel BS.50. 500 = 500 Reaktionen x 50 µl Haltbarkeit: 12 Monate Lagerung bei
Nur für Forschungszwecke Datenblatt Artikel BS.50.020 = 20 Reaktionen x 50 µl Artikel BS.50.100 = 100 Reaktionen x 50 µl Artikel BS.50. 500 = 500 Reaktionen x 50 µl Haltbarkeit: 12 Monate Lagerung bei
Die Entwicklung der Keimbahn. wahrend der Embryogenese von Caenorhabditis elegans
 Die Entwicklung der Keimbahn wahrend der Embryogenese von Caenorhabditis elegans Von der Fakultat fur Lebenswissenschaften der Technischen Universitat Carolo-Wilhelmina zu Braunschweig zur Erlangung des
Die Entwicklung der Keimbahn wahrend der Embryogenese von Caenorhabditis elegans Von der Fakultat fur Lebenswissenschaften der Technischen Universitat Carolo-Wilhelmina zu Braunschweig zur Erlangung des
Transkriptionelle Repression als molekulare Grundlage der anti-inflammatorischen Wirkung von Glucocorticoiden
 Forschungszentrum Karlsruhe Technik und Umwelt Wissenschaftliche Berichte FZKA 6087 Transkriptionelle Repression als molekulare Grundlage der anti-inflammatorischen Wirkung von Glucocorticoiden Stefanie
Forschungszentrum Karlsruhe Technik und Umwelt Wissenschaftliche Berichte FZKA 6087 Transkriptionelle Repression als molekulare Grundlage der anti-inflammatorischen Wirkung von Glucocorticoiden Stefanie
Inhaltsverzeichnis. Zusammenfassung 1. Summary 4. 1 Einleitung Das Prostatakarzinom Entstehung und Bedeutung von Lymphknotenmetastasen 9
 Inhaltsverzeichnis Zusammenfassung 1 Summary 4 1 Einleitung 7 1.1 Das Prostatakarzinom 7 1.2 Entstehung und Bedeutung von Lymphknotenmetastasen 9 1.2.1 Das Lymphsystem 9 1.2.2 Lymphknotenmetastasierung
Inhaltsverzeichnis Zusammenfassung 1 Summary 4 1 Einleitung 7 1.1 Das Prostatakarzinom 7 1.2 Entstehung und Bedeutung von Lymphknotenmetastasen 9 1.2.1 Das Lymphsystem 9 1.2.2 Lymphknotenmetastasierung
Untersuchungen zur differenziellen Genexpression im ZNS von Noradrenalintransporter-Knockout- und Wildtyp-Mäusen
 Untersuchungen zur differenziellen Genexpression im ZNS von Noradrenalintransporter-Knockout- und Wildtyp-Mäusen Inaugural-Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen
Untersuchungen zur differenziellen Genexpression im ZNS von Noradrenalintransporter-Knockout- und Wildtyp-Mäusen Inaugural-Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen
Stammzellenmanipulation. Stammzellen können in Zellkultur manipuliert werden
 Stammzellenmanipulation Hämatopoietische Stammzellen können gebraucht werden um kranke Zellen mit gesunden zu ersetzen (siehe experiment bestrahlte Maus) Epidermale Stammzellpopulationen können in Kultur
Stammzellenmanipulation Hämatopoietische Stammzellen können gebraucht werden um kranke Zellen mit gesunden zu ersetzen (siehe experiment bestrahlte Maus) Epidermale Stammzellpopulationen können in Kultur
Expression der genetischen Information Skript: Kapitel 5
 Prof. A. Sartori Medizin 1. Studienjahr Bachelor Molekulare Zellbiologie FS 2013 12. März 2013 Expression der genetischen Information Skript: Kapitel 5 5.1 Struktur der RNA 5.2 RNA-Synthese (Transkription)
Prof. A. Sartori Medizin 1. Studienjahr Bachelor Molekulare Zellbiologie FS 2013 12. März 2013 Expression der genetischen Information Skript: Kapitel 5 5.1 Struktur der RNA 5.2 RNA-Synthese (Transkription)
Zeitaufgelöste Fluoreszenzmessung zur Verwendung bei der Realtime-PCR
 Zeitaufgelöste Fluoreszenzmessung zur Verwendung bei der Realtime-PCR Nils Scharke, inano Abstract Die Verwendung neuartiger Seltenerd-Komplexe in Verbindung mit zeitaufgelöster Fluoreszenzmessung liefert
Zeitaufgelöste Fluoreszenzmessung zur Verwendung bei der Realtime-PCR Nils Scharke, inano Abstract Die Verwendung neuartiger Seltenerd-Komplexe in Verbindung mit zeitaufgelöster Fluoreszenzmessung liefert
1. PCR Polymerase-Kettenreaktion
 entechnische Verfahren 1. PCR Polymerase-Kettenreaktion Die PCR (engl. Polymerase Chain Reaction) ist eine Methode, um die DNA zu vervielfältigen, ohne einen lebenden Organismus, wie z.b. Escherichia coli
entechnische Verfahren 1. PCR Polymerase-Kettenreaktion Die PCR (engl. Polymerase Chain Reaction) ist eine Methode, um die DNA zu vervielfältigen, ohne einen lebenden Organismus, wie z.b. Escherichia coli
Modul 3.4 Grundlagen der Tumorerkrankungen Sommersemester 2004 Vorlesung Einführung in die Onkologie Organisation der Module
 Modul 3.4 Grundlagen der Tumorerkrankungen Sommersemester 2004 Vorlesung 1 14.06.04 Einführung in die Onkologie Organisation der Module Klinische Onkologie Zweithäufigste Todesursache Tendenz steigend
Modul 3.4 Grundlagen der Tumorerkrankungen Sommersemester 2004 Vorlesung 1 14.06.04 Einführung in die Onkologie Organisation der Module Klinische Onkologie Zweithäufigste Todesursache Tendenz steigend
erläutern Eigenschaften des genetischen Codes und charakterisieren mit dessen Hilfe Experimentelle Entschlüsselung (SF)
") Schulinterner Kernlehrplan Biologie Q1 : Genetik Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen 1.1 Vom Gen zum Genprodukt Wiederholung - DNA und Replikation Aufgaben DNA und Replikation
Schulinterner Kernlehrplan Biologie Q1 : Genetik Inhaltsfelder Schwerpunkt Basiskonzept Konkretisierte Kompetenzen 1.1 Vom Gen zum Genprodukt Wiederholung - DNA und Replikation Aufgaben DNA und Replikation
Familienanamnese, Tumorlokalisation, Tumorabrenzung, Tumorgröße, TNM-Stadien
 Prognosefaktoren beim Die Prognose dieser Studie untersucht die Dauer des rezidivfreien Intervalls und die Gesamtüberlebenszeit. Folgende Prognosefaktoren wurden berücksichtigt: 1.klinische Daten 2. histopathologischer
Prognosefaktoren beim Die Prognose dieser Studie untersucht die Dauer des rezidivfreien Intervalls und die Gesamtüberlebenszeit. Folgende Prognosefaktoren wurden berücksichtigt: 1.klinische Daten 2. histopathologischer
1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und
 Welche besondere Art von Nukleotiden wird verwendet und") 10. Methoden 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und welche Funktion haben diese bei der Sequenzierungsreaktion?
10. Methoden 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet und welche Funktion haben diese bei der Sequenzierungsreaktion?
Chronische myeloische Leukämie. Chronische Phase
 Chronische myeloische Leukämie Chronische Phase Zytologie Prof. Dr. med. Roland Fuchs Prof. Dr. med. Tim Brümmendorf Medizinische Klinik IV Zytogenetik Molekulargenetik Prof. Dr. med. Detlef Haase Zentrum
Chronische myeloische Leukämie Chronische Phase Zytologie Prof. Dr. med. Roland Fuchs Prof. Dr. med. Tim Brümmendorf Medizinische Klinik IV Zytogenetik Molekulargenetik Prof. Dr. med. Detlef Haase Zentrum
Inhaltsverzeichnis. 1 Einleitung... 1
 Inhaltsverzeichnis 1 Einleitung... 1 1.1 G-Protein-gekoppelte Rezeptoren... 3 1.1.1 Klassifizierung und Struktur von G-Protein-gekoppelten Rezeptoren... 6 1.1.2 Purinerge Rezeptoren... 12 1.1.3 Pharmazeutische
Inhaltsverzeichnis 1 Einleitung... 1 1.1 G-Protein-gekoppelte Rezeptoren... 3 1.1.1 Klassifizierung und Struktur von G-Protein-gekoppelten Rezeptoren... 6 1.1.2 Purinerge Rezeptoren... 12 1.1.3 Pharmazeutische
Die DNA Replikation. Exakte Verdopplung des genetischen Materials. Musterstrang. Neuer Strang. Neuer Strang. Eltern-DNA-Doppelstrang.
 Die DNA Replikation Musterstrang Neuer Strang Eltern-DNA-Doppelstrang Neuer Strang Musterstrang Exakte Verdopplung des genetischen Materials Die Reaktion der DNA Polymerase 5`-Triphosphat Nächstes Desoxyribonucleosidtriphosphat
Die DNA Replikation Musterstrang Neuer Strang Eltern-DNA-Doppelstrang Neuer Strang Musterstrang Exakte Verdopplung des genetischen Materials Die Reaktion der DNA Polymerase 5`-Triphosphat Nächstes Desoxyribonucleosidtriphosphat
Gentransfer in höhere Eukaryonten
 63 Gentransfer in höhere Eukaryonten von Florian Rüker, Wien Mit Hilfe der Methoden der DNA-Rekombination ist es möglich geworden, eine Vielzahl von Genen zu isolieren und zu charakterisieren. Die funktionelle
63 Gentransfer in höhere Eukaryonten von Florian Rüker, Wien Mit Hilfe der Methoden der DNA-Rekombination ist es möglich geworden, eine Vielzahl von Genen zu isolieren und zu charakterisieren. Die funktionelle
Angewandte Zellbiologie
 Angewandte Zellbiologie Bt-BZ01: Pflanzenzellen als Bioreaktoren 8 CP (VL + Pr) MENDEL (VL) HÄNSCH (Pr) Bt-BZ02: Zellbiologie der Tiere I 8 CP (VL + Pr) BUCHBERGER WINTER (VL) VAUTI (Pr) Bt-BZ03: Zellbiologie
Angewandte Zellbiologie Bt-BZ01: Pflanzenzellen als Bioreaktoren 8 CP (VL + Pr) MENDEL (VL) HÄNSCH (Pr) Bt-BZ02: Zellbiologie der Tiere I 8 CP (VL + Pr) BUCHBERGER WINTER (VL) VAUTI (Pr) Bt-BZ03: Zellbiologie
Weitergabe genetischer Information: DNA-Replikation Beispiel: Escherichia coli.
 Weitergabe genetischer Information: DNA-Replikation Beispiel: Escherichia coli. zirkuläres bakterielles Chromosom Replikation (Erstellung einer identischen Kopie des genetischen Materials) MPM 1 DNA-Polymerasen
Weitergabe genetischer Information: DNA-Replikation Beispiel: Escherichia coli. zirkuläres bakterielles Chromosom Replikation (Erstellung einer identischen Kopie des genetischen Materials) MPM 1 DNA-Polymerasen
Stellungnahme zur Ernährung von Tumorpatienten auf der Grundlage der "Anti TKTL1 - Diät"
 Deutsche Krebsgesellschaft Stellungnahme zur Ernährung von Tumorpatienten auf der Grundlage der "Anti TKTL1 - Diät" Berlin (18. März 2010) - Tumorpatienten wird derzeit ein neues Ernährungsprinzip mit
Deutsche Krebsgesellschaft Stellungnahme zur Ernährung von Tumorpatienten auf der Grundlage der "Anti TKTL1 - Diät" Berlin (18. März 2010) - Tumorpatienten wird derzeit ein neues Ernährungsprinzip mit
Unterrichtsmaterialien in digitaler und in gedruckter Form. Auszug aus: Lernwerkstatt: Genetik & Vererbung. Das komplette Material finden Sie hier:
 Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: : Genetik & Vererbung Das komplette Material finden Sie hier: Download bei School-Scout.de Inhalt Vorwort Seite 4 Einleitung Seite
Unterrichtsmaterialien in digitaler und in gedruckter Form Auszug aus: : Genetik & Vererbung Das komplette Material finden Sie hier: Download bei School-Scout.de Inhalt Vorwort Seite 4 Einleitung Seite
Zweigbibliothek Medizin
 Sächsische Landesbibliothek - Staats- und Universitätsbibliothek Dresden (SLUB) Zweigbibliothek Medizin Diese Hochschulschrift finden Sie original in Printform zur Ausleihe in der Zweigbibliothek Medizin
Sächsische Landesbibliothek - Staats- und Universitätsbibliothek Dresden (SLUB) Zweigbibliothek Medizin Diese Hochschulschrift finden Sie original in Printform zur Ausleihe in der Zweigbibliothek Medizin
Kontrolle des Zellzyklus: Checkpoints. kann an bestimmten Kontrollpunkten gestoppt werden
 Kontrolle des Zellzyklus: Checkpoints kann an bestimmten Kontrollpunkten gestoppt werden G2 checkpoint komplette DNA Replikation? sind DNA Schäden vorhanden? ist die Zelle groß genug? ist die Umgebung
Kontrolle des Zellzyklus: Checkpoints kann an bestimmten Kontrollpunkten gestoppt werden G2 checkpoint komplette DNA Replikation? sind DNA Schäden vorhanden? ist die Zelle groß genug? ist die Umgebung
Eukaryontische DNA-Bindedomänen
 1. Viele eukaryotische (und auch prokaryotische) Transkriptionsfaktoren besitzen eine DNA-bindende Domäne, die an eine ganz bestimmte DNA- Sequenz binden kann. Aufgrund von Ähnlichkeiten in der Struktur
1. Viele eukaryotische (und auch prokaryotische) Transkriptionsfaktoren besitzen eine DNA-bindende Domäne, die an eine ganz bestimmte DNA- Sequenz binden kann. Aufgrund von Ähnlichkeiten in der Struktur
Untersuchungen zur Wirkung des Ginkgo biloba-extraktts EGb 761 auf die Proteinaggregation in der Huntington-Krankheit
 Untersuchungen zur Wirkung des Ginkgo biloba-extraktts EGb 761 auf die Proteinaggregation in der Huntington-Krankheit Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften am Fachbereich
Untersuchungen zur Wirkung des Ginkgo biloba-extraktts EGb 761 auf die Proteinaggregation in der Huntington-Krankheit Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften am Fachbereich
Inhalte unseres Vortrages
 Inhalte unseres Vortrages Vorstellung der beiden paper: Germ line transmission of a disrupted ß2 mirkroglobulin gene produced by homologous recombination in embryonic stem cells ß2 Mikroglobulin deficient
Inhalte unseres Vortrages Vorstellung der beiden paper: Germ line transmission of a disrupted ß2 mirkroglobulin gene produced by homologous recombination in embryonic stem cells ß2 Mikroglobulin deficient
RNA-Regulationsmechanismen: RNA Interferenz
 RNA-Regulationsmechanismen: RNA Interferenz Vorlesung System-Biophysik 19. Dez. 2008 Literatur Martens: BIOspektrum 4/02 8. Jahrgang M. Kuhlmann: Biol. Unserer Zeit Nr.3 (2004), S. 142. Genregulation durch
RNA-Regulationsmechanismen: RNA Interferenz Vorlesung System-Biophysik 19. Dez. 2008 Literatur Martens: BIOspektrum 4/02 8. Jahrgang M. Kuhlmann: Biol. Unserer Zeit Nr.3 (2004), S. 142. Genregulation durch
Alternative Methoden der RNA-Analyse
 Alternative Methoden der RNA-Analyse In diesem Versuch wurde die Northern Blot Hybridisierung zur Analyse isolierter mrna eingesetzt. Mit dieser Technik können Größe und Menge einer spezifischen RNA bestimmt
Alternative Methoden der RNA-Analyse In diesem Versuch wurde die Northern Blot Hybridisierung zur Analyse isolierter mrna eingesetzt. Mit dieser Technik können Größe und Menge einer spezifischen RNA bestimmt
Trisomie 21: Entstehung - Folgen - Lebenserwartung/-standard - Auftretenswahrscheinlichkeit
 Naturwissenschaft Lorenz Wächter Trisomie 21: Entstehung - Folgen - Lebenserwartung/-standard - Auftretenswahrscheinlichkeit Studienarbeit Inhaltsverzeichnis 1. Begriffsbestimmung 1 2. Entstehung...1
Naturwissenschaft Lorenz Wächter Trisomie 21: Entstehung - Folgen - Lebenserwartung/-standard - Auftretenswahrscheinlichkeit Studienarbeit Inhaltsverzeichnis 1. Begriffsbestimmung 1 2. Entstehung...1
Raumfahrtmedizin. darauf hin, dass es auch einen Zusammenhang zwischen Knochenabbau, Bluthochdruck und Kochsalzzufuhr gibt. 64 DLR NACHRICHTEN 113
 Raumfahrtmedizin Gibt es einen Die geringe mechanische Belastung der unteren Extremitäten von Astronauten im All ist eine wesentliche Ursache für den Knochenabbau in Schwerelosigkeit. Gleichzeitig haben
Raumfahrtmedizin Gibt es einen Die geringe mechanische Belastung der unteren Extremitäten von Astronauten im All ist eine wesentliche Ursache für den Knochenabbau in Schwerelosigkeit. Gleichzeitig haben
Synthese von eukaryotischen RNA-Modifikationen
 Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Synthese von eukaryotischen RNA-Modifikationen und Quantifizierung nicht kanonischer
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Synthese von eukaryotischen RNA-Modifikationen und Quantifizierung nicht kanonischer
Karyotypanalysen durch Chromosomen-Färbungen an HeLa-Zelllinien
 Karyotypanalysen durch Chromosomen-Färbungen an HeLa-Zelllinien Sonja Prohaska 9808503 A441 sonja@bioinf.uni-leipzig.de 1 Einleitung HeLa-Zellen sind Zervixkarzinom-Zellen, die ihre begrenzte Teilbarkeit,
Karyotypanalysen durch Chromosomen-Färbungen an HeLa-Zelllinien Sonja Prohaska 9808503 A441 sonja@bioinf.uni-leipzig.de 1 Einleitung HeLa-Zellen sind Zervixkarzinom-Zellen, die ihre begrenzte Teilbarkeit,
4. ERGEBNISSE. 4.2. Untersuchung des umgelagerten IgH Gens in Hodgkinzelllinien
 36 4. ERGEBNISSE 4.1. Immunglobulin Gentranskripte in Hodgkinzelllinien Mit Hilfe der RT PCR untersuchten wir die Expression umgelagerter Ig Gene in den Hodgkinzelllinien L1236, L428, L591 und KM-H2 sowie
36 4. ERGEBNISSE 4.1. Immunglobulin Gentranskripte in Hodgkinzelllinien Mit Hilfe der RT PCR untersuchten wir die Expression umgelagerter Ig Gene in den Hodgkinzelllinien L1236, L428, L591 und KM-H2 sowie
Molekulare Mechanismen der Signaltransduktion. 06 - Kartierung des AXR1 Gens + early auxin-induced genes Folien: http://tinyurl.
 Molekulare Mechanismen der Signaltransduktion 06 - Kartierung des AXR1 Gens + early auxin-induced genes Folien: http://tinyurl.com/modul-mms bisheriges Modell auxin auxin AXR1 auxin response AXR1 potentieller
Molekulare Mechanismen der Signaltransduktion 06 - Kartierung des AXR1 Gens + early auxin-induced genes Folien: http://tinyurl.com/modul-mms bisheriges Modell auxin auxin AXR1 auxin response AXR1 potentieller
Molekulare Diagnostik in der Zytologie
 Molekulare Diagnostik in der Zytologie Nicole Pawlaczyk Karsten Neumann Institut für Pathologie Pneumologische Onkologie Lungenkarzinom häufigste krebsbedingte Todesursache für beide Geschlechter dritthäufigste
Molekulare Diagnostik in der Zytologie Nicole Pawlaczyk Karsten Neumann Institut für Pathologie Pneumologische Onkologie Lungenkarzinom häufigste krebsbedingte Todesursache für beide Geschlechter dritthäufigste
Das Transketolase Protein TKTL1 ist in Ovarialkarzinomen überexprimiert
 Földi M 1., zur Hausen A 2., Jäger M 1., Bau L 1., Kretz O 3., Watermann D 1., Gitsch G 1., Stickeler E 1., Coy JF 4. Das Transketolase Protein TKTL1 ist in Ovarialkarzinomen überexprimiert 1 Universitäts-Frauenklinik
Földi M 1., zur Hausen A 2., Jäger M 1., Bau L 1., Kretz O 3., Watermann D 1., Gitsch G 1., Stickeler E 1., Coy JF 4. Das Transketolase Protein TKTL1 ist in Ovarialkarzinomen überexprimiert 1 Universitäts-Frauenklinik
DNA Sequenzierung. Transkriptionsstart bestimmen PCR
 10. Methoden DNA Sequenzierung Transkriptionsstart bestimmen PCR 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet
10. Methoden DNA Sequenzierung Transkriptionsstart bestimmen PCR 1. Erklären Sie das Prinzip der Sanger Sequenzierung. Klären Sie dabei folgende Punkte: a) Welche besondere Art von Nukleotiden wird verwendet
Molekulargenetischer Nachweis und Genotypisierung von HPV
 Molekulargenetischer Nachweis und Genotypisierung von HPV Dr. Sabine Sussitz-Rack Institut für Labordiagnostik und Mikrobiologie Vorstand: Prim. Prof. DDr. Pranav Sinha Humanes Papillomavirus HPV HPV ist
Molekulargenetischer Nachweis und Genotypisierung von HPV Dr. Sabine Sussitz-Rack Institut für Labordiagnostik und Mikrobiologie Vorstand: Prim. Prof. DDr. Pranav Sinha Humanes Papillomavirus HPV HPV ist
1. Stammbaum einer Familie, in der Mukoviszidose aufgetreten ist.
 Die Prüfungsarbeit besteht aus drei zu bearbeitenden Teilen Aufgabe I Aufgabe II A oder II B Aufgabe III A oder III B I Aufgabe I: Humangenetik / klassische Genetik / Molekulargenetik Mukoviszidose Mukoviszidose,
Die Prüfungsarbeit besteht aus drei zu bearbeitenden Teilen Aufgabe I Aufgabe II A oder II B Aufgabe III A oder III B I Aufgabe I: Humangenetik / klassische Genetik / Molekulargenetik Mukoviszidose Mukoviszidose,
4. Diskussion. 4.1. Polymerase-Kettenreaktion
 59 4. Diskussion Bei der Therapie maligner Tumoren stellt die Entwicklung von Kreuzresistenz gegen Zytostatika ein ernstzunehmendes Hindernis dar. Im wesentlich verantwortlich für die so genannte Multidrug
59 4. Diskussion Bei der Therapie maligner Tumoren stellt die Entwicklung von Kreuzresistenz gegen Zytostatika ein ernstzunehmendes Hindernis dar. Im wesentlich verantwortlich für die so genannte Multidrug
Produktkatalog 2010. Molekularbiologische Reagenzien
 Produktion, Vertrieb und Serviceleistung im Bereich der Molekularbiologie und Medizin Produktkatalog 2010 Molekularbiologische Reagenzien Molegene GmbH Bienenweg 28 35764 Sinn Tel. 02772-570952 Fax 02772-570945
Produktion, Vertrieb und Serviceleistung im Bereich der Molekularbiologie und Medizin Produktkatalog 2010 Molekularbiologische Reagenzien Molegene GmbH Bienenweg 28 35764 Sinn Tel. 02772-570952 Fax 02772-570945
Biochemie Tutorium 9. RNA, Transkription
 Biochemie Tutorium 9 RNA, Transkription IMPP-Gegenstandskatalog 3 Genetik 3.1 Nukleinsäuren 3.1.1 Molekulare Struktur, Konformationen und Funktionen der Desoxyribonukleinsäure (DNA); Exon, Intron 3.1.2
Biochemie Tutorium 9 RNA, Transkription IMPP-Gegenstandskatalog 3 Genetik 3.1 Nukleinsäuren 3.1.1 Molekulare Struktur, Konformationen und Funktionen der Desoxyribonukleinsäure (DNA); Exon, Intron 3.1.2
Telomersequenzen unterschiedlicher Organismen. Organismus Telomersequenz Referenz
 3 1 EINLEITUNG 1.1 Telomere und Telomerase 1.1.1 Telomere Als Telomere werden die Endabschnitte eukaryontischer Chromosomen bezeichnet. Der Name Telomere setzt sich aus den griechischen Worten telos (=
3 1 EINLEITUNG 1.1 Telomere und Telomerase 1.1.1 Telomere Als Telomere werden die Endabschnitte eukaryontischer Chromosomen bezeichnet. Der Name Telomere setzt sich aus den griechischen Worten telos (=
Der Blick in die Telomere
 Quelle: picture alliance/bildagentur-online Der Blick in die Was verraten sie über unser Leben? Ist der Mensch geboren, so fängt er an zu sterben. Diese Volkweisheit mag zwar etwas deprimierend erscheinen,
Quelle: picture alliance/bildagentur-online Der Blick in die Was verraten sie über unser Leben? Ist der Mensch geboren, so fängt er an zu sterben. Diese Volkweisheit mag zwar etwas deprimierend erscheinen,
Kaninchen: 1 cagattgaggagagcaccaccatcgtcacctcgcagttggcggaggtcagcgcagccgag 60
 Ergebnisse 6 3. Ergebnisse 3. Charakterisierung der zelllinienspezifischen cdn Die Entwicklung zweier Zelllinien, des epithelialen Trophoblasten und des pluripotenten Embryoblasten, ist der erste Differenzierungsschritt
Ergebnisse 6 3. Ergebnisse 3. Charakterisierung der zelllinienspezifischen cdn Die Entwicklung zweier Zelllinien, des epithelialen Trophoblasten und des pluripotenten Embryoblasten, ist der erste Differenzierungsschritt
Ergebnisse. I. Identifizierung intrazellulärer Interaktionspartner. Ergebnisse I. Identifizierung intrazellulärer Interaktionspartner
 Ergebnisse I. Identifizierung intrazellulärer Interaktionspartner Es sollten mithilfe des Yeast-Two-Hybrid-Systems Proteine identifiziert werden, die mit der zytoplasmatischen Domäne von CEACAM1-4L aus
Ergebnisse I. Identifizierung intrazellulärer Interaktionspartner Es sollten mithilfe des Yeast-Two-Hybrid-Systems Proteine identifiziert werden, die mit der zytoplasmatischen Domäne von CEACAM1-4L aus
Antwort: 2.Uracil. Antwort: 2. durch Wasserstoffverbindungen. Adenin, Cystein und Guanin kommen alle in der RNA und DNA vor.
 Antwort: 2.Uracil Adenin, Cystein und Guanin kommen alle in der RNA und DNA vor. Thymin kommt nur in der DNA vor; Uracil nimmt seinen Platz in den RNA- Molekülen ein. Antwort: 2. durch Wasserstoffverbindungen
Antwort: 2.Uracil Adenin, Cystein und Guanin kommen alle in der RNA und DNA vor. Thymin kommt nur in der DNA vor; Uracil nimmt seinen Platz in den RNA- Molekülen ein. Antwort: 2. durch Wasserstoffverbindungen
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr
 Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
Klausur zum Modul Molekularbiologie ILS, SS 2010 Freitag 6. August 10:00 Uhr Name: Matrikel-Nr.: Code Nummer: Bitte geben Sie Ihre Matrikel-Nr. und Ihren Namen an. Die Code-Nummer erhalten Sie zu Beginn
DNA versus RNA. RNA Instabilität
 DNA versus RNA DNA stellt den eigentlichen Speicher genetischer Information dar, während RNA als Informationsüberträger und katalytisch in der Proteinbiosynthese agiert. Warum dient DNA und nicht RNA als
DNA versus RNA DNA stellt den eigentlichen Speicher genetischer Information dar, während RNA als Informationsüberträger und katalytisch in der Proteinbiosynthese agiert. Warum dient DNA und nicht RNA als
Rekombinante Antikorperfragmente fur die. Zoonosediagnostik
 Rekombinante Antikorperfragmente fur die Zoonosediagnostik Von der Fakultat fur Lebenswissenschaften der Technischen Universitat Carolo-Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors
Rekombinante Antikorperfragmente fur die Zoonosediagnostik Von der Fakultat fur Lebenswissenschaften der Technischen Universitat Carolo-Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors
β2-microglobulin deficient mice lack CD4-8+cytolytic T cells
 β2-microglobulin deficient mice lack CD4-8+cytolytic T cells Mäuse mit einem Knock-out bezüglich ß-Microglobulin sind nicht in der Lage CD4-8+ cytotoxische T-Zellen zu bilden Nature,Vol 344, 19. April
β2-microglobulin deficient mice lack CD4-8+cytolytic T cells Mäuse mit einem Knock-out bezüglich ß-Microglobulin sind nicht in der Lage CD4-8+ cytotoxische T-Zellen zu bilden Nature,Vol 344, 19. April
Die perfekte Kombination. Effektive Biomarker-Kombination zur Früherkennung von hochgradigen Vorstufen des Zervixkarzinoms
 Früherkennung des Zervixkarzinoms: Die perfekte Kombination. Effektive Biomarker-Kombination zur Früherkennung von hochgradigen Vorstufen des Zervixkarzinoms p16 PLUS Ki-67 in einem einzelnen Test CLARITY
Früherkennung des Zervixkarzinoms: Die perfekte Kombination. Effektive Biomarker-Kombination zur Früherkennung von hochgradigen Vorstufen des Zervixkarzinoms p16 PLUS Ki-67 in einem einzelnen Test CLARITY
5 Zusammenfassung und Schlussfolgerung
 5 Zusammenfassung und Schlussfolgerung Einleitung In der Schwangerschaft vollziehen sich Veränderungen des Kohlenhydratstoffwechsels im Sinne einer Insulinresistenz sowie eines Anstieges der Blutfettwerte.
5 Zusammenfassung und Schlussfolgerung Einleitung In der Schwangerschaft vollziehen sich Veränderungen des Kohlenhydratstoffwechsels im Sinne einer Insulinresistenz sowie eines Anstieges der Blutfettwerte.
Phylogenetisches Praktikum im ITZ 08.02.2010-19.02.2010
 Phylogenetisches Praktikum im ITZ 08.02.2010-19.02.2010 Zellzyklus- und neuronale Gene in Trichoplax adhaerens S. Sagasser Praktikanten: Patrick Reinke und Jan Kleveman Betreuerin: Karolin von der Chevallerie
Phylogenetisches Praktikum im ITZ 08.02.2010-19.02.2010 Zellzyklus- und neuronale Gene in Trichoplax adhaerens S. Sagasser Praktikanten: Patrick Reinke und Jan Kleveman Betreuerin: Karolin von der Chevallerie
Inhalt. 3 Das Werkzeug... 47 3.1 Restriktionsenzyme... 47 3.2 Gele... 58 3.2.1 Agarosegele... 58
 Inhalt 1 Was ist denn Molekularbiologie, bitteschön?...................... 1 1.1 Das Substrat der Molekularbiologie, oder: Molli-World für Anfänger.... 2 1.2 Was brauche ich zum Arbeiten?..................................
Inhalt 1 Was ist denn Molekularbiologie, bitteschön?...................... 1 1.1 Das Substrat der Molekularbiologie, oder: Molli-World für Anfänger.... 2 1.2 Was brauche ich zum Arbeiten?..................................
Strahleninduzierte Apoptose bei ESRT im Mausmodell
 Strahleninduzierte Apoptose bei ESRT im Mausmodell Die Apoptose ist von vielen ineinandergreifenden Mechanismen abhängig, in deren Regulationsmittelpunkt die Caspasen als ausführende Cysteinproteasen stehen.
Strahleninduzierte Apoptose bei ESRT im Mausmodell Die Apoptose ist von vielen ineinandergreifenden Mechanismen abhängig, in deren Regulationsmittelpunkt die Caspasen als ausführende Cysteinproteasen stehen.
Molekulargenetische und biochemische Untersuchungen zur Pathogenese der Spinalen Muskelatrophie mit Atemnot Typ 1 (SMARD1)
") Molekulargenetische und biochemische Untersuchungen zur Pathogenese der Spinalen Muskelatrophie mit Atemnot Typ 1 (SMARD1) Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften
Molekulargenetische und biochemische Untersuchungen zur Pathogenese der Spinalen Muskelatrophie mit Atemnot Typ 1 (SMARD1) Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften
Station 1. Analyse von strahleninduzierten DNA-Schäden durch Gel-Elektrophorese
 Station 1 Analyse von strahleninduzierten DNA-Schäden durch Gel-Elektrophorese 1 I. Vorinformationen An der GSI wurde eine weltweit neuartige Krebstherapie mit Ionenstrahlen entwickelt. Dazu werden in
Station 1 Analyse von strahleninduzierten DNA-Schäden durch Gel-Elektrophorese 1 I. Vorinformationen An der GSI wurde eine weltweit neuartige Krebstherapie mit Ionenstrahlen entwickelt. Dazu werden in
Drexler G.A. 1, Derer A 1, Dirks W.G. 2, Hable V. 3, Greubel C. 3, Burgdorfer C. 3, Dollinger G. 3, Du G. 4, Friedl A.A. 1
 Nutzung von bi-cistronischen Vektoren für die Beobachtung der Rekrutierung von Signal- und Reparaturproteinen an DNA-Schäden nach Ionen- Mikrobestrahlung durch Live-Cell Imaging Drexler G.A. 1, Derer A
Nutzung von bi-cistronischen Vektoren für die Beobachtung der Rekrutierung von Signal- und Reparaturproteinen an DNA-Schäden nach Ionen- Mikrobestrahlung durch Live-Cell Imaging Drexler G.A. 1, Derer A
EINFLUSS DER ZELLMEMBRAN UND EINIGER ZELLAKTOREN AUF DEN ALTERUNGSPROZESS
 EINFLUSS DER ZELLMEMBRAN UND EINIGER ZELLAKTOREN AUF DEN ALTERUNGSPROZESS Der Kern steuert die Proteinsynthese durch Synthese der Boten-RNA (mrna) gemäß den von der DNA gelieferten Informationen Die mrna
EINFLUSS DER ZELLMEMBRAN UND EINIGER ZELLAKTOREN AUF DEN ALTERUNGSPROZESS Der Kern steuert die Proteinsynthese durch Synthese der Boten-RNA (mrna) gemäß den von der DNA gelieferten Informationen Die mrna
1. Einleitung S. 1 1.1. Zelladhäsionsmoleküle S. 1 1.2. Die Immunglobulin-Superfamilie S. 2. 1.2.1. Das neurale Zelladhäsionsmolekül NCAM S.
 Inhaltsverzeichnis 1. Einleitung S. 1 1.1. Zelladhäsionsmoleküle S. 1 1.2. Die Immunglobulin-Superfamilie S. 2 1.2.1. Das neurale Zelladhäsionsmolekül NCAM S. 4 1.2.1.1. Molekulare Struktur von NCAM S.
Inhaltsverzeichnis 1. Einleitung S. 1 1.1. Zelladhäsionsmoleküle S. 1 1.2. Die Immunglobulin-Superfamilie S. 2 1.2.1. Das neurale Zelladhäsionsmolekül NCAM S. 4 1.2.1.1. Molekulare Struktur von NCAM S.
Das Paper von heute. Samuel Grimm & Jan Kemna
 Das Paper von heute Samuel Grimm & Jan Kemna Bisheriges Modell Was bereits bekannt war - TIR1 ist an Auxinantwort (Zellteilung, Elongation, Differenzierung) beteiligt, im selben Signalweg wie AXR1 - TIR1
Das Paper von heute Samuel Grimm & Jan Kemna Bisheriges Modell Was bereits bekannt war - TIR1 ist an Auxinantwort (Zellteilung, Elongation, Differenzierung) beteiligt, im selben Signalweg wie AXR1 - TIR1
Rekombinante Antikörper
 Frank Breitling und Stefan Dübel 2008 AGI-Information Management Consultants May be used for personal purporses only or by libraries associated to dandelon.com network. Rekombinante Antikörper Technische
Frank Breitling und Stefan Dübel 2008 AGI-Information Management Consultants May be used for personal purporses only or by libraries associated to dandelon.com network. Rekombinante Antikörper Technische
RNA-guided editing of bacterial genomes using CRISPR-Cas systems
 RNA-guided editing of bacterial genomes using CRISPR-Cas systems Wenyan Jiang, David Bikard, David Cox, Feng Zhang, and Luciano A. Marraffini Nature Biotechnology, 31, 233 239, 2013 Nadja Kleisch Biotechnologie,
RNA-guided editing of bacterial genomes using CRISPR-Cas systems Wenyan Jiang, David Bikard, David Cox, Feng Zhang, and Luciano A. Marraffini Nature Biotechnology, 31, 233 239, 2013 Nadja Kleisch Biotechnologie,
Chromosomen & Populationsgenetik (1)
") Übungsblatt Molekularbiologie und Genetik für Studierende der Bioinformatik II 1 Name des Studierenden: Datum: 1 Karyogramme Chromosomen & Populationsgenetik (1) Bestimmen Sie den Karyotyp der folgenden
Übungsblatt Molekularbiologie und Genetik für Studierende der Bioinformatik II 1 Name des Studierenden: Datum: 1 Karyogramme Chromosomen & Populationsgenetik (1) Bestimmen Sie den Karyotyp der folgenden
Die Östrogenrezeptorgen- (ESR1) Amplifikation beim Mammakarzinom: Ein neuer prädiktiver Marker?
 Amplifikation beim Mammakarzinom: Ein neuer prädiktiver Marker?") Die Östrogenrezeptorgen- (ESR1) Amplifikation beim Mammakarzinom: Ein neuer prädiktiver Marker? Frederik Holst Institut für Pathologie Universitätsklinikum Hamburg-Eppendorf Genamplifikation normale Kopiezahl
Die Östrogenrezeptorgen- (ESR1) Amplifikation beim Mammakarzinom: Ein neuer prädiktiver Marker? Frederik Holst Institut für Pathologie Universitätsklinikum Hamburg-Eppendorf Genamplifikation normale Kopiezahl
Genetische Ursachen der Krebsentstehung Molekularbiologie menschlicher Tumoren
 Genetische Ursachen der Krebsentstehung Molekularbiologie menschlicher Tumoren Literatur Molekulare Onkologie Entstehung und Progression maligner Tumoren C. Wagener Thieme Verlag Was ist Krebs? Zellzyklus
Genetische Ursachen der Krebsentstehung Molekularbiologie menschlicher Tumoren Literatur Molekulare Onkologie Entstehung und Progression maligner Tumoren C. Wagener Thieme Verlag Was ist Krebs? Zellzyklus
Mutation ist nicht gleich Mutation
 Arbeitsblatt 8: Mutation ist nicht gleich Mutation Mutationen können eine Zelle aus dem physiologischen Gleichgewicht bringen, weil sich dadurch beispielsweise die Menge oder Aktivität produzierter Proteine
Arbeitsblatt 8: Mutation ist nicht gleich Mutation Mutationen können eine Zelle aus dem physiologischen Gleichgewicht bringen, weil sich dadurch beispielsweise die Menge oder Aktivität produzierter Proteine
5 Signalwegdefekte in malignen Erkrankungen
 5 Signalwegdefekte in malignen Erkrankungen Aufgrund der häufig geringen prognostischen Relevanz konventioneller klinischpathologischer Parameter besteht erheblicher Bedarf an neuen diagnostischen Verfahren,
5 Signalwegdefekte in malignen Erkrankungen Aufgrund der häufig geringen prognostischen Relevanz konventioneller klinischpathologischer Parameter besteht erheblicher Bedarf an neuen diagnostischen Verfahren,
Cornel Mülhardt. Genomics. 6. Auflaqe. Spektrum k - / l AKADEMISCHER VERLAG
 I Cornel Mülhardt Genomics 6. Auflaqe Spektrum k - / l AKADEMISCHER VERLAG Inhalt 1 Was ist denn "Molekularbiologie", bitteschön? 1 1.1 Das Substrat der Molekularbiologie, oder: Molli-World für Anfänger...
I Cornel Mülhardt Genomics 6. Auflaqe Spektrum k - / l AKADEMISCHER VERLAG Inhalt 1 Was ist denn "Molekularbiologie", bitteschön? 1 1.1 Das Substrat der Molekularbiologie, oder: Molli-World für Anfänger...
Lsd1 weist Stammzellen im Mausembryo den richtigen Weg
 Powered by Seiten-Adresse: https://www.gesundheitsindustriebw.de/de/fachbeitrag/aktuell/lsd1-weist-stammzellen-immausembryo-den-richtigen-weg/ Lsd1 weist Stammzellen im Mausembryo den richtigen Weg Die
Powered by Seiten-Adresse: https://www.gesundheitsindustriebw.de/de/fachbeitrag/aktuell/lsd1-weist-stammzellen-immausembryo-den-richtigen-weg/ Lsd1 weist Stammzellen im Mausembryo den richtigen Weg Die
Zusätzliche Behandlungs möglichkeiten für Ihre Patienten
 Zusätzliche Behandlungs möglichkeiten für Ihre Patienten Über FoundationOne Bei dem validierten umfassenden Tumorprofil von FoundationOne wird die gesamte codierende Sequenz von 315 krebsassoziierten Genen
Zusätzliche Behandlungs möglichkeiten für Ihre Patienten Über FoundationOne Bei dem validierten umfassenden Tumorprofil von FoundationOne wird die gesamte codierende Sequenz von 315 krebsassoziierten Genen
Molekularbiologie/ Genomics
 Cornel Mülhardt Molekularbiologie/ Genomics 5. Auflage ELSEVIER SPEKTRUM AKADEMISCHER VERLAG Spektrum k-zlakademischer VERLAG Inhalt 1 Was ist denn "Molekularbiologie", bitteschön? 1 1.1 Das Substrat der
Cornel Mülhardt Molekularbiologie/ Genomics 5. Auflage ELSEVIER SPEKTRUM AKADEMISCHER VERLAG Spektrum k-zlakademischer VERLAG Inhalt 1 Was ist denn "Molekularbiologie", bitteschön? 1 1.1 Das Substrat der
4. Genetische Mechanismen bei Bakterien
 4. Genetische Mechanismen bei Bakterien 4.1 Makromoleküle und genetische Information Aufbau der DNA Phasen des Informationsflusses Vergleich der Informationsübertragung bei Pro- und Eukaryoten 4.2 Struktur
4. Genetische Mechanismen bei Bakterien 4.1 Makromoleküle und genetische Information Aufbau der DNA Phasen des Informationsflusses Vergleich der Informationsübertragung bei Pro- und Eukaryoten 4.2 Struktur
1. Definition und Mechanismen
 Zusammenfassung 1. Definition und Mechanismen Epigenetik (von griechisch epi- über ) bezeichnet erbliche Veränderungen in der Genexpression, die nicht von Veränderungen in der DNA Sequenz (Mutationen)
Zusammenfassung 1. Definition und Mechanismen Epigenetik (von griechisch epi- über ) bezeichnet erbliche Veränderungen in der Genexpression, die nicht von Veränderungen in der DNA Sequenz (Mutationen)
- 1 - am 19.02.2003 im Großen Vortragssaal des Ärztehauses der Ärztekammer Niedersachsen, Hamburger Allee 20, 30175 Hannover
 - 1 - Vortrag über Harnblasenkarzinom Neue Aspekte in Diagnostik und Therapie (Oberarzt Dr.med. Machtens) - Neue molekularpathologische Untersuchungen zur Früherkennung und Verlaufskontrolle des Harnblasenkarzinom
- 1 - Vortrag über Harnblasenkarzinom Neue Aspekte in Diagnostik und Therapie (Oberarzt Dr.med. Machtens) - Neue molekularpathologische Untersuchungen zur Früherkennung und Verlaufskontrolle des Harnblasenkarzinom
Anabole Prozesse in der Zelle
 Anabole Prozesse in der Zelle DNA Vermehrung RNA Synthese Protein Synthese Protein Verteilung in der Zelle Ziel: Zellteilung (Wachstum) und Differenzierung (Aufgabenteilung im Organismus). 2016 Struktur
Anabole Prozesse in der Zelle DNA Vermehrung RNA Synthese Protein Synthese Protein Verteilung in der Zelle Ziel: Zellteilung (Wachstum) und Differenzierung (Aufgabenteilung im Organismus). 2016 Struktur
Labortechniken (2) Amplifikationskurve (linear linear) Realtime-PCR
 Amplifikationskurve (linear linear) Realtime-PCR") Realtime PCR Quantitative PCR Prinzip: Nachweis der PCR-Produkte in Echtzeit und Quantifizierung anhand eines fluoreszenten Reporters R Q Taq-Polymerase Synthese- und Nuklease-Einheit R=Reporter, Q=Quencher
Realtime PCR Quantitative PCR Prinzip: Nachweis der PCR-Produkte in Echtzeit und Quantifizierung anhand eines fluoreszenten Reporters R Q Taq-Polymerase Synthese- und Nuklease-Einheit R=Reporter, Q=Quencher
Abbildungsverzeichnis
 I INHALTSVERZEICHNIS Abkürzungen VI Abbildungsverzeichnis VIII I. Einleitung 1. Neurone und Axonwachstum 1 2. Oligodendrozyten und Myelin 3 3. Das Proteolipid Protein (PLP) 6 4. Mutationen im PLP-Gen und
I INHALTSVERZEICHNIS Abkürzungen VI Abbildungsverzeichnis VIII I. Einleitung 1. Neurone und Axonwachstum 1 2. Oligodendrozyten und Myelin 3 3. Das Proteolipid Protein (PLP) 6 4. Mutationen im PLP-Gen und
Von der DNA zum Eiweißmolekül Die Proteinbiosynthese. Ribosom
 Von der DNA zum Eiweißmolekül Die Proteinbiosynthese Ribosom Wiederholung: DNA-Replikation und Chromosomenkondensation / Mitose Jede Zelle macht von Teilung zu Teilung einen Zellzyklus durch, der aus einer
Von der DNA zum Eiweißmolekül Die Proteinbiosynthese Ribosom Wiederholung: DNA-Replikation und Chromosomenkondensation / Mitose Jede Zelle macht von Teilung zu Teilung einen Zellzyklus durch, der aus einer
Heterologe Expression einer funktionellen Domäne des nikotinischen Acetylcholinrezeptors
 Heterologe Expression einer funktionellen Domäne des nikotinischen Acetylcholinrezeptors Inauguraldissertation zur Erlangung der Doktorwürde am Fachbereich Biologie/Chemie/Pharmazie der Freien Universität
Heterologe Expression einer funktionellen Domäne des nikotinischen Acetylcholinrezeptors Inauguraldissertation zur Erlangung der Doktorwürde am Fachbereich Biologie/Chemie/Pharmazie der Freien Universität
Humane mesenchymale Stammzellen (HMSC): Differenzierung und Fusion mit anderen Zellarten
: Differenzierung und Fusion mit anderen Zellarten") Humane mesenchymale Stammzellen (HMSC): Differenzierung und Fusion mit anderen Zellarten Dissertation Zur Erlangung des Akademischen Grades doctor rerum naturalium (Dr. rer. nat.) vorgelegt der Mathematisch-Naturwissenschaftlichen-Technischen
Humane mesenchymale Stammzellen (HMSC): Differenzierung und Fusion mit anderen Zellarten Dissertation Zur Erlangung des Akademischen Grades doctor rerum naturalium (Dr. rer. nat.) vorgelegt der Mathematisch-Naturwissenschaftlichen-Technischen